Advanced Manufacturing of Tucatinib Intermediates: A Pd-Catalyzed Route for Scalable Production
The pharmaceutical landscape for HER2-positive breast cancer treatment has been significantly advanced by the development of small molecule tyrosine kinase inhibitors, with Tucatinib standing out as a pivotal therapeutic agent. As detailed in the recent intellectual property disclosure CN114230568B, a novel preparation method has been engineered to address the longstanding inefficiencies in synthesizing this critical active pharmaceutical ingredient (API). This patent outlines a robust four-step synthetic pathway that leverages innovative catalytic systems to achieve superior selectivity and yield. For global procurement leaders and R&D directors, understanding this methodology is crucial, as it represents a shift towards more sustainable and cost-effective pharmaceutical intermediates manufacturing. The core of this invention lies in its ability to streamline the construction of the complex molecular architecture of Tucatinib, specifically optimizing the final coupling stages to minimize byproduct formation.
Traditionally, the synthesis of kinase inhibitors like Tucatinib has been plagued by convoluted reaction sequences that introduce unnecessary operational risks and cost burdens. Conventional routes often rely on a strategy where a nitro group is first reduced to an amine, followed by a halogenation step to enable subsequent coupling. This approach is inherently flawed because it increases the total number of unit operations, thereby compounding material losses at each stage. Furthermore, the presence of multiple reactive sites in intermediate structures can lead to non-specific substitutions, generating difficult-to-remove impurities that compromise the purity profile required for clinical applications. These limitations necessitate rigorous purification protocols, often involving resource-intensive column chromatography, which acts as a bottleneck in commercial scale-up of complex kinase inhibitors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
The legacy manufacturing processes for HER2 inhibitors typically suffer from low atom economy and poor step efficiency. In standard protocols, the conversion of nitro-aromatic precursors into the final amine-coupled product requires distinct reduction and activation steps. Each additional step introduces potential points of failure, such as incomplete conversion or over-reaction, which complicates the impurity profile. Moreover, the use of harsh reagents for halogenation can degrade sensitive functional groups elsewhere in the molecule, leading to significant yield erosion. From a supply chain perspective, these inefficiencies translate to longer lead times and higher volatility in raw material consumption. The reliance on chromatographic purification further exacerbates these issues, as it requires large volumes of organic solvents and specialized silica media, driving up both the environmental footprint and the cost of goods sold (COGS) for the final high-purity HER2 inhibitor intermediates.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN114230568B introduces a paradigm shift by utilizing a direct catalytic amination strategy in the critical fourth step. Instead of reducing the nitro group prematurely, the inventors employ a sophisticated palladium catalyst system, specifically Pd(acac)2 paired with a specialized Ligand L, to facilitate the coupling reaction directly on the nitro-containing scaffold. This innovation ensures that the reaction site remains singular and highly specific, effectively bypassing the side reactions associated with free amines. The result is a streamlined process that maintains the integrity of the molecular backbone while achieving exceptional conversion rates. By eliminating the need for intermediate isolation and activation, this route significantly shortens the production timeline and enhances the overall throughput for cost reduction in pharmaceutical manufacturing.
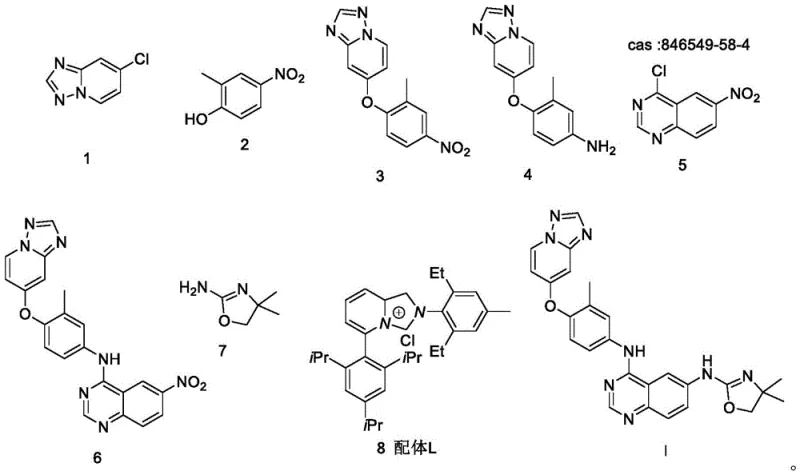
Mechanistic Insights into Pd(acac)2-Catalyzed Amination
The mechanistic elegance of this synthesis lies in the precise orchestration of the palladium catalytic cycle during the final amination step. The use of Pd(acac)2 (Palladium(II) acetylacetonate) in conjunction with Ligand L creates a highly active catalytic species capable of oxidative addition into the carbon-halogen bond of the quinazoline core without interfering with the nitro group on the adjacent ring. This chemoselectivity is paramount; in traditional nucleophilic aromatic substitution, the nitro group might undergo unwanted reduction or side reactions under the necessary thermal conditions. However, the ligand-modified palladium center stabilizes the transition state, lowering the activation energy for the desired C-N bond formation while kinetically inhibiting competing pathways. This ensures that the reaction proceeds with high fidelity, preserving the oxidation state of the nitro group until it is no longer a liability, or allowing it to remain as a stable spectator throughout the coupling event.
Furthermore, the choice of base and solvent system plays a critical role in managing the impurity profile. The protocol specifies the use of K3PO4·3H2O in dioxane at a controlled temperature of 90°C. This mild basic environment is sufficient to deprotonate the incoming amine nucleophile without causing hydrolysis of the sensitive triazolopyridine or oxazole moieties. The result is a reaction mixture that is remarkably clean, facilitating the downstream purification process. By minimizing the formation of des-halo byproducts or homocoupling impurities, the process ensures that the crude product already meets stringent quality thresholds. This mechanistic control is what allows the manufacturers to forego complex chromatographic separations, relying instead on simple crystallization or slurry techniques to achieve the required purity specifications for reliable API intermediate supplier standards.
How to Synthesize Tucatinib Efficiently
The implementation of this synthetic route requires strict adherence to the optimized reaction parameters defined in the patent to ensure reproducibility and safety. The process begins with the formation of the ether linkage via nucleophilic substitution, followed by a selective nitro reduction, and then the assembly of the quinazoline core. The final and most critical step involves the palladium-catalyzed coupling, which demands precise stoichiometry and atmosphere control. Operators must ensure that the reaction vessel is thoroughly purged with nitrogen to prevent catalyst deactivation by oxygen. The detailed standardized operating procedures for each transformation, including specific addition rates and quenching protocols, are essential for maintaining the high yields reported in the experimental data. For a comprehensive breakdown of the exact operational steps required to execute this chemistry in a GMP environment, please refer to the technical guide below.
- Perform nucleophilic substitution between chloro-triazolopyridine and methyl-nitrophenol using K2CO3 in DMF at 100°C to form the ether linkage.
- Reduce the nitro group of the intermediate using Pd/C and hydrogen gas in methanol at 20-30°C to generate the aniline derivative.
- Couple the aniline with chloro-quinazoline using K2CO3 in DMF at 80°C to construct the core kinase inhibitor scaffold.
- Execute the final amination using Pd(acac)2, Ligand L, and K3PO4·3H2O in dioxane at 90°C to install the oxazole moiety without nitro reduction.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers tangible strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the purification workflow. By replacing column chromatography with slurry purification (beating) using common solvent systems like petroleum ether and ethyl acetate, the process eliminates a major cost center. Chromatography is not only expensive in terms of media and solvent consumption but also limits batch size due to column capacity constraints. Removing this bottleneck allows for true batch processing, which is essential for meeting the high-volume demands of the global oncology market. This operational shift directly contributes to cost reduction in pharmaceutical manufacturing by lowering variable costs and increasing equipment utilization rates.
- Cost Reduction in Manufacturing: The economic impact of this route is driven by the elimination of expensive purification media and the reduction of solvent waste. Traditional methods often require vast quantities of silica gel and eluents, which constitute a significant portion of the COGS. By utilizing a precipitation-based purification strategy, the material costs are slashed, and the waste disposal burden is lightened. Additionally, the high yield of each step—reported consistently above 90% in the patent examples—means that less raw material is required to produce a kilogram of final API. This efficiency translates into substantial savings on starting materials, particularly for the specialized heterocyclic building blocks which can be cost-prohibitive if used inefficiently.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by complex syntheses that rely on hard-to-source reagents or unstable intermediates. This route mitigates those risks by utilizing commercially available starting materials and robust catalysts. The stability of the intermediates allows for potential storage between steps if necessary, providing flexibility in production scheduling. Moreover, the shortened reaction sequence reduces the total manufacturing cycle time, enabling faster response to market demand fluctuations. For a reliable API intermediate supplier, this means the ability to commit to tighter delivery windows and maintain safety stock levels without incurring excessive inventory carrying costs.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces new safety and environmental challenges, but this methodology is inherently designed for expansion. The reaction conditions, such as temperatures ranging from 80°C to 100°C, are well within the operational limits of standard glass-lined or stainless steel reactors, avoiding the need for cryogenic cooling or high-pressure autoclaves. The reduction in solvent usage and the avoidance of silica waste align with green chemistry principles, simplifying regulatory compliance and environmental permitting. This makes the technology highly attractive for facilities aiming to reduce their carbon footprint while expanding capacity for commercial scale-up of complex kinase inhibitors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Tucatinib synthesis pathway. These insights are derived directly from the experimental data and claims presented in patent CN114230568B, providing a factual basis for decision-making. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer or licensing agreements. The answers reflect the specific advantages of the Pd-catalyzed route over legacy methods, focusing on yield, purity, and operational simplicity.
Q: What is the key innovation in patent CN114230568B for Tucatinib synthesis?
A: The patent introduces a novel fourth step using Pd(acac)2 and Ligand L to perform direct amination on a nitro-containing intermediate. This avoids the conventional multi-step reduction-halogenation sequence, resulting in fewer reaction sites, higher selectivity, and improved overall yield.
Q: How does this method improve purification compared to traditional routes?
A: Traditional methods often require expensive and time-consuming column chromatography. This patented process utilizes simple slurry purification (beating) with petroleum ether/ethyl acetate mixtures after each step, significantly simplifying post-treatment and reducing production costs.
Q: Is this synthetic route suitable for large-scale commercial manufacturing?
A: Yes, the route is designed for industrial operability. It uses commercially available starting materials, avoids hazardous reagents where possible, and achieves high yields (over 90% in key steps) with robust temperature controls (75°C-100°C), making it ideal for metric-ton scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tucatinib Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for life-saving oncology treatments like Tucatinib. Our technical team has extensively analyzed the innovations presented in CN114230568B and possesses the expertise to implement this advanced Pd-catalyzed amination strategy at an industrial level. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to full-scale manufacturing is seamless. Our facilities are equipped with state-of-the-art rigorous QC labs capable of monitoring the specific impurity profiles associated with kinase inhibitor synthesis, guaranteeing that every batch meets stringent purity specifications required by global regulatory bodies.
We invite pharmaceutical partners to collaborate with us to leverage this cost-effective and high-yield manufacturing technology. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to request specific COA data and route feasibility assessments that demonstrate how our optimized production capabilities can secure your supply chain for HER2 inhibitor intermediates. Let us help you accelerate your drug development timeline with a reliable and economically viable supply partner.