Advanced Capecitabine Manufacturing: High-Purity Synthesis and Commercial Scalability
Advanced Capecitabine Manufacturing: High-Purity Synthesis and Commercial Scalability
The pharmaceutical industry continuously demands more efficient, cost-effective, and high-purity synthetic routes for critical oncology therapeutics like capecitabine. Patent CN103601777A introduces a transformative preparation method that addresses longstanding challenges in the synthesis of this vital antimetabolite. By fundamentally re-engineering the crystallization and purification stages, this technology converts difficult-to-handle oily intermediates into stable solids, thereby enabling precise stoichiometric control in downstream reactions. This breakthrough not only elevates the final product purity to an exceptional 99.96% but also drastically simplifies the manufacturing workflow, offering a compelling value proposition for global supply chains seeking reliable API intermediate suppliers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of capecitabine has been plagued by inefficiencies in the handling of key intermediates, particularly during the formation of 2',3'-bis-O-acetyl-5'-deoxy-5-fluoro-N-(pentyloxycarbonyl)cytidine. Traditional protocols frequently rely on anhydrous sodium sulfate for dehydration purposes, a step that paradoxically leads to significant product entrapment and a marked reduction in overall mass yield. Furthermore, prior art crystallization techniques often utilize ethyl acetate and n-hexane systems, which have proven inadequate for effectively refining the crude product. In many documented cases, capecitabine exhibits poor solubility characteristics in ethyl acetate, resulting in disordered crystallization patterns, low recovery rates, and a final purity profile that offers negligible improvement over the crude state, rendering the purification step economically and technically insignificant.
The Novel Approach
The innovative methodology disclosed in the patent data circumvents these historical bottlenecks through a strategic shift in solvent systems and physical state management. Instead of struggling with oily residues, the new process employs a specific mixture of isopropyl ether and acetonitrile to induce crystallization of the intermediate, successfully converting the substance from an intractable oil into a free-flowing white powder. This physical transformation is critical for industrial operations as it allows for accurate weighing and feeding into the subsequent deprotection reaction. Additionally, the final purification stage abandons the problematic ethyl acetate approach in favor of a dichloromethane and toluene system, which effectively solubilizes impurities while precipitating high-purity capecitabine, thereby overcoming the disadvantages of bad crystal state and low yield inherent in legacy technologies.
Mechanistic Insights into Acylation and Deprotection Kinetics
The core of this synthetic strategy lies in the precise control of reaction conditions during the acylation and subsequent hydrolysis phases. As illustrated in the reaction scheme below, the process begins with the acylation of 2',3'-bis-O-acetyl-5'-deoxy-5-fluorocytidine using n-amyl chloroformate. This reaction is conducted in aprotic solvents such as dichloromethane or acetonitrile in the presence of a basifier like pyridine or triethylamine. Maintaining the reaction temperature strictly between -5°C and -10°C is paramount to minimizing side reactions and ensuring the selective formation of the carbamate linkage at the N4 position without compromising the integrity of the acetyl protecting groups on the sugar moiety.
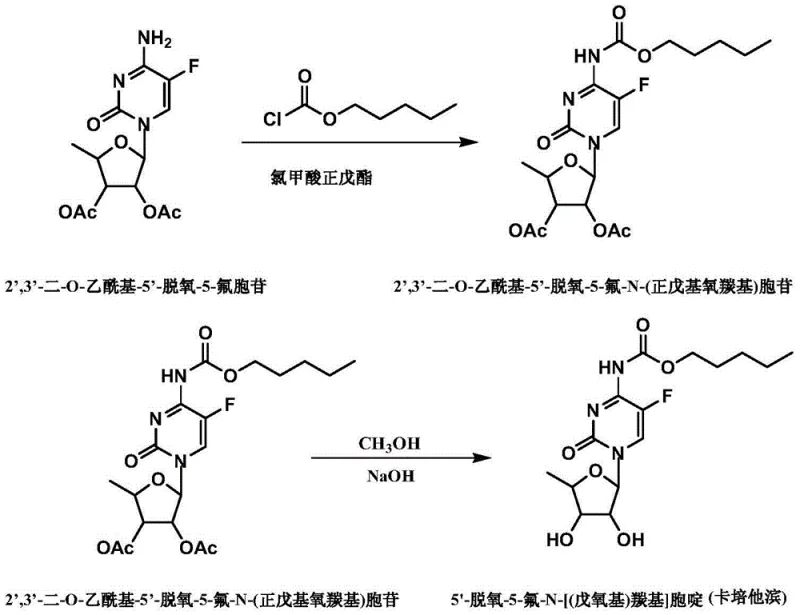
Following the isolation of the intermediate, the mechanism shifts to alkaline hydrolysis for deprotection. The intermediate is dissolved in methanol and treated with a sodium hydroxide solution at controlled low temperatures, typically around -3°C to -7°C. This step cleaves the acetyl groups to reveal the hydroxyl functionalities essential for biological activity. A crucial aspect of this mechanism is the subsequent pH adjustment to a narrow range of 4.5 to 4.6 using concentrated hydrochloric acid. This precise acidification ensures the stability of the glycosidic bond while facilitating the extraction of the product into the organic phase, effectively separating it from inorganic salts and polar byproducts generated during the neutralization process.
How to Synthesize Capecitabine Efficiently
The implementation of this patented route requires strict adherence to solvent ratios and temperature profiles to maximize the transition from oily intermediates to crystalline solids. The process is divided into three distinct operational units: the formation and crystallization of the carbamate intermediate, the alkaline deprotection to form the crude active pharmaceutical ingredient, and the final recrystallization to meet pharmacopeial standards. Operators must ensure that the initial acylation is quenched properly and that the concentration of the organic phase is managed carefully before the addition of the anti-solvent system. Detailed standardized operating procedures for each unit operation are provided in the technical guide below to ensure reproducibility and safety.
- Perform acylation of 2',3'-bis-O-acetyl-5'-deoxy-5-fluorocytidine with n-amyl chloroformate in dichloromethane at -5 to -10°C, followed by crystallization using isopropyl ether and acetonitrile.
- Conduct alkaline hydrolysis of the intermediate in methanol using sodium hydroxide, adjust pH to 4.5-4.6, and extract with dichloromethane to obtain crude capecitabine.
- Refine the crude product by dissolving in dichloromethane and crystallizing with toluene to remove impurities and achieve final purity specifications.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis route translates directly into enhanced operational efficiency and reduced total cost of ownership. The elimination of the anhydrous sodium sulfate drying step is not merely a technical tweak but a significant process intensification measure. By removing the need for solid-phase drying agents, the process reduces the volume of solid waste generated and eliminates the filtration losses associated with separating these agents from the product mixture. This streamlining results in a substantial increase in mass yield, meaning less raw material is required to produce the same amount of finished goods, driving down the variable cost per kilogram of production significantly.
- Cost Reduction in Manufacturing: The economic impact of this technology is driven by the drastic simplification of the purification workflow. Traditional methods often require multiple recrystallization cycles to achieve acceptable purity, consuming vast amounts of solvents and energy. In contrast, this novel approach achieves a purity of 99.96% through a single, highly effective crystallization step using dichloromethane and toluene. The ability to convert the intermediate into a solid form also prevents the loss of material that typically occurs when handling viscous oils, ensuring that nearly all synthesized material proceeds to the final step, thereby optimizing the return on investment for expensive starting materials.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by batch-to-batch variability, particularly when dealing with unstable oily intermediates that degrade or vary in potency. By stabilizing the intermediate as a crystalline solid, this method ensures consistent quality and precise dosing for the subsequent reaction. This consistency reduces the risk of batch failures and off-specification products, which are major causes of supply delays. Furthermore, the use of common, commercially available solvents like toluene and methanol ensures that the supply chain is not vulnerable to shortages of exotic or highly regulated reagents, securing the long-term availability of the product.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, the reduction in solvent usage and solid waste generation aligns with modern green chemistry principles. The process avoids the generation of large quantities of sulfate-contaminated waste, simplifying effluent treatment and reducing disposal costs. The robustness of the crystallization process means that scaling from pilot plant quantities to multi-ton commercial production can be achieved with minimal re-optimization. The predictable physical properties of the intermediates allow for the use of standard industrial equipment, reducing the capital expenditure required for facility upgrades and accelerating the time to market for generic or biosimilar versions of the drug.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this capecitabine synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on how this method outperforms legacy processes in terms of purity, yield, and operational simplicity. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer.
Q: How does the new crystallization method improve capecitabine purity?
A: The patented method replaces traditional ethyl acetate/n-hexane systems with a dichloromethane and toluene combination. This specific solvent pair effectively removes impurities that co-crystallize in older methods, allowing the final product purity to reach 99.96%.
Q: What are the cost advantages of avoiding anhydrous sodium sulfate in this process?
A: Traditional methods often rely on anhydrous sodium sulfate for dehydration, which can lead to significant product loss and lower yields. By eliminating this step and utilizing direct crystallization from the organic phase, the process significantly improves mass yield and reduces material waste.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the process is designed for scalability. The conversion of the intermediate from an oil to a stable white powder solid allows for precise feeding in subsequent steps, ensuring consistent reaction stoichiometry and facilitating reliable scale-up from pilot to commercial tonnage.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Capecitabine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high purity and yield demonstrated in patent literature can be consistently replicated on an industrial scale. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify that every batch of capecitabine meets the highest global regulatory standards, providing our partners with absolute confidence in product quality.
We invite pharmaceutical companies and contract manufacturers to collaborate with us to leverage this advanced synthesis route for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential partners to contact us directly to obtain specific COA data from our recent batches and to discuss route feasibility assessments that can help optimize your production costs and secure a stable supply of this critical oncology intermediate.