Advanced Deprotection Strategy for High-Purity Losartan Intermediates and Commercial Scale-Up
Advanced Deprotection Strategy for High-Purity Losartan Intermediates and Commercial Scale-Up
The global demand for angiotensin II receptor antagonists continues to drive rigorous innovation in the synthesis of key antihypertensive agents like Losartan Potassium. A critical bottleneck in the commercial production of this active pharmaceutical ingredient (API) has historically been the control of specific genotoxic or structurally related impurities, particularly dimeric byproducts. Patent CN108047208B, published in March 2022, introduces a transformative methodology for reducing losartan dimer impurities through a precisely controlled acid-hydrolysis protocol. This technical breakthrough addresses the longstanding issue where losartan intermediates, specifically the condensation compounds, are prone to dimerization under acidic conditions, complicating downstream purification. By optimizing reaction parameters such as pH, temperature, and solvent systems, this method achieves dimer levels below 0.2%, and in optimized scenarios, below 0.1%, representing a significant leap forward for manufacturers seeking a reliable pharmaceutical intermediates supplier capable of delivering ultra-high purity standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
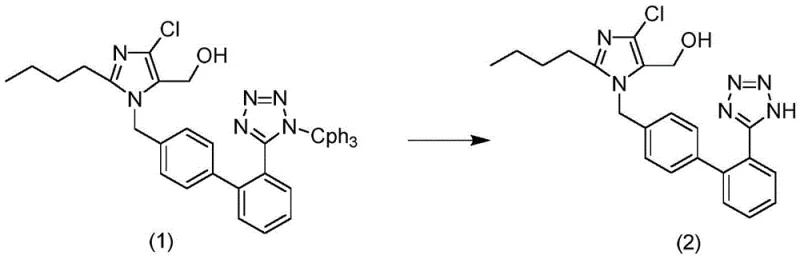
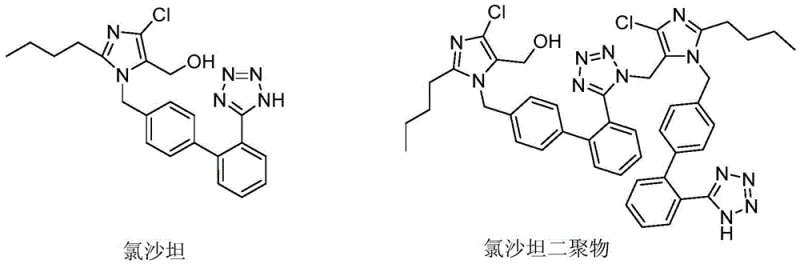
Traditional synthetic routes for Losartan often involve the deprotection of a trityl-group protected tetrazole intermediate using strong acidic conditions. However, the chemical nature of the biphenyl-tetrazole scaffold makes it inherently susceptible to electrophilic attack or condensation reactions when exposed to protons in solution. In conventional processes, the acidic environment required to cleave the protecting group simultaneously catalyzes the formation of dimer impurities, where two losartan molecules link together, typically through the tetrazole or imidazole rings. Removing these dimers is notoriously difficult because their physicochemical properties closely resemble the target molecule, necessitating multiple, energy-intensive recrystallization steps or complex chromatographic separations. These heavy refining procedures not only consume vast amounts of solvents and manpower but also result in substantial product loss, drastically reducing the overall process yield and rendering the final cost of goods sold (COGS) uncompetitive in the generic pharmaceutical market.
The Novel Approach
The methodology disclosed in CN108047208B circumvents these pitfalls by decoupling the deprotection efficiency from the dimerization rate through kinetic control. Instead of harsh, uncontrolled acidification, the process utilizes a moderated acidic environment with a pH strictly maintained between 1 and 5, preferably between 2 and 4. This narrow window is sufficient to hydrolyze the trityl protecting group from the losartan condensation compound but is mild enough to suppress the kinetics of dimer formation. Furthermore, the reaction is conducted at moderate temperatures ranging from 10°C to 40°C, preventing thermal acceleration of side reactions. The use of specific organic solvents like tetrahydrofuran or isopropanol further stabilizes the transition state, ensuring that the conversion to the free tetrazole form proceeds cleanly. This strategic adjustment allows for a direct crystallization after workup, eliminating the need for repetitive purification cycles and securing a high-purity product in a single operational sequence.
Mechanistic Insights into Acid-Catalyzed Deprotection and Impurity Suppression
To fully appreciate the value of this process for R&D directors, one must understand the delicate balance of the reaction mechanism. The starting material, the losartan condensation compound, typically contains a triphenylmethyl (trityl) group protecting the tetrazole nitrogen. The cleavage of this C-N bond requires protonation, yet the resulting free tetrazole anion/cation equilibrium in acidic media is the precursor to dimerization. The patent suggests that by controlling the acid concentration (preferably 1 to 4 mol/L hydrochloric acid) and the addition temperature (10-20°C), the local concentration of reactive cationic species is kept low. This prevents the intermolecular coupling that leads to the dimer structure shown in the comparative analysis. The reaction time of 8 to 20 hours ensures complete conversion of the starting material without pushing the system into a thermodynamic equilibrium that favors the dimer. This mechanistic precision is what allows the process to achieve yields exceeding 95% while maintaining impurity profiles that would otherwise require extensive downstream processing.
Furthermore, the workup procedure plays a pivotal role in the final purity profile. After the acidic reaction, the system is cooled and basified to a pH of 10-14. This step serves a dual purpose: it neutralizes the acid to stop the reaction instantly and converts the losartan into its water-soluble salt form, allowing for the separation of organic-soluble non-polar impurities. The subsequent evaporation of the organic solvent and re-acidification of the aqueous phase induces crystallization of the free acid form of losartan. Because the dimer impurities remain in solution or are excluded from the crystal lattice due to the high purity of the mother liquor established in the first step, the final solid obtained is exceptionally pure. This crystallization-driven purification is far more scalable and cost-effective than chromatographic methods, making it ideal for industrial applications.
How to Synthesize Losartan Efficiently
Implementing this synthesis route requires strict adherence to the parameter windows defined in the patent to ensure reproducibility at scale. The process begins with the dissolution of the losartan condensation compound in a selected organic solvent, followed by the controlled addition of acid to initiate deprotection. The reaction mixture is then stirred under温和 conditions before undergoing a phase-switch workup involving basification, solvent removal, and final acid-induced crystallization. This streamlined workflow minimizes unit operations and maximizes throughput. For detailed operational parameters, stoichiometry, and safety considerations, please refer to the standardized synthesis guide below.
- Dissolve the losartan condensation compound in an organic solvent such as tetrahydrofuran or isopropanol, maintaining a mass-to-volume ratio between 2.0 and 10.0 ml/g.
- Add acid (e.g., hydrochloric acid) at 10-30°C to adjust the system pH to 1-5, then stir at 10-40°C for 8-20 hours to complete the deprotection reaction.
- Cool the mixture, adjust to alkaline pH (10-14) with base, evaporate the solvent, add water, filter, re-acidify the filtrate to pH 2-5, and crystallize to obtain pure losartan.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology translates directly into enhanced operational efficiency and risk mitigation. The primary advantage lies in the drastic simplification of the purification train. By preventing the formation of dimer impurities at the source rather than removing them post-synthesis, manufacturers can eliminate entire stages of refining, such as repeated recrystallizations or column chromatography. This reduction in processing steps leads to substantial cost savings in terms of solvent consumption, energy usage for heating and cooling, and labor hours. Additionally, the high yield reported in the patent examples, consistently hovering around 94% to 96%, means that less raw material is wasted, directly improving the atom economy and reducing the cost per kilogram of the final API intermediate.
- Cost Reduction in Manufacturing: The elimination of heavy refining steps significantly lowers the variable costs associated with production. Traditional methods often suffer from yield erosion due to multiple purification cycles; in contrast, this novel approach secures high recovery rates in a single crystallization event. By utilizing common, commodity-grade solvents like tetrahydrofuran and isopropanol, along with standard mineral acids, the process avoids the need for exotic or expensive reagents. This accessibility of raw materials ensures that cost reduction in pharmaceutical intermediates manufacturing is sustainable and not reliant on volatile specialty chemical markets.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to a more reliable supply chain. The process operates at mild temperatures (10-40°C) and atmospheric pressure, reducing the dependency on specialized high-pressure or cryogenic equipment that can be prone to maintenance downtime. The tolerance for a range of solvents (THF, IPA, Methanol) provides flexibility; if one solvent faces supply constraints, the process can be adapted to another without compromising quality. This flexibility reduces lead time for high-purity pharmaceutical intermediates and ensures continuity of supply even during market fluctuations.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, the method is highly favorable. The reduction in solvent volume and the avoidance of complex separation techniques lower the facility's environmental footprint, aiding in compliance with increasingly stringent waste disposal regulations. The simplicity of the workup—essentially involving evaporation and filtration—is easily transferable from pilot plant to commercial scale. This ease of commercial scale-up of complex pharmaceutical intermediates allows manufacturers to rapidly respond to market demand surges without the lengthy validation periods typically associated with modifying complex purification protocols.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this impurity control strategy. These insights are derived directly from the experimental data and claims within the patent documentation, providing a clear framework for evaluating the technology's fit within existing production lines. Understanding these specifics is crucial for technical teams assessing the feasibility of technology transfer.
Q: What is the primary impurity challenge in Losartan synthesis addressed by this patent?
A: The primary challenge is the formation of dimer impurities, which occur easily under acidic conditions during the synthesis and solution states. These dimers are difficult to remove via standard refining, often requiring heavy purification processes that reduce yield and increase costs.
Q: Which solvents are compatible with this high-yield purification method?
A: The patent specifies several effective organic solvents including tetrahydrofuran (THF), isopropanol, and methanol. Tetrahydrofuran is noted as the preferred solvent for achieving optimal reaction kinetics and impurity control.
Q: What yield and purity levels can be expected from this process?
A: Experimental data in the patent demonstrates yields ranging from 92% to 96%. Crucially, the dimer impurity content is consistently controlled below 0.2%, with optimized examples achieving levels as low as 0.08%.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Losartan Supplier
At NINGBO INNO PHARMCHEM, we recognize that the difference between a viable project and a commercial success often lies in the purity and consistency of the starting materials. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the laboratory efficiencies demonstrated in patents like CN108047208B are faithfully reproduced in our manufacturing facilities. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs equipped with state-of-the-art analytical instrumentation, guaranteeing that every batch of Losartan intermediate meets the exacting standards required by global regulatory bodies.
We invite you to collaborate with us to leverage this advanced purification technology for your supply chain. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this low-impurity route can optimize your bottom line. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate why we are the preferred partner for high-value pharmaceutical intermediates.