Mastering Sugammadex Sodium Impurity Synthesis for Global Pharmaceutical Compliance
The pharmaceutical landscape for neuromuscular blocking reversal agents is dominated by Sugammadex Sodium, a modified gamma-cyclodextrin that has revolutionized anesthesia recovery protocols globally. As regulatory scrutiny intensifies around the world, the identification and control of process-related impurities have become paramount for maintaining market authorization and patient safety. Patent CN110818817A introduces a pivotal advancement in this domain by detailing a robust preparation method for a specific diphenyl phosphine oxide derivative impurity, which serves as an essential reference standard for quality control. This technical breakthrough addresses the critical need for characterizing toxicologically non-characteristic impurities that may arise during the complex multi-step synthesis of the active pharmaceutical ingredient. By establishing a reliable pathway to generate this specific analog, manufacturers can ensure their analytical methods are sufficiently sensitive to detect even trace levels of potential degradants or side products. 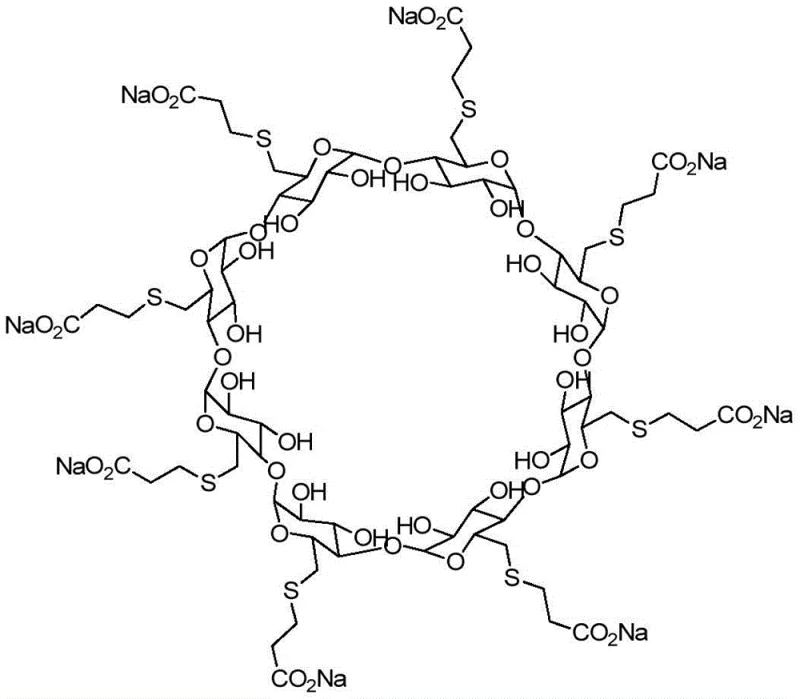
The structural complexity of Sugammadex Sodium necessitates a deep understanding of its potential degradation pathways and synthetic by-products. The molecule features eight carboxyethyl thio ether groups attached to the gamma-cyclodextrin backbone, creating numerous sites for potential chemical variation during manufacturing. The impurity described in the referenced patent represents a scenario where diphenylphosphine, often used in the iodination steps of the API synthesis, reacts to form a stable phosphine oxide derivative. Having access to a authenticated sample of this impurity is not merely a regulatory checkbox but a fundamental requirement for validating the specificity of HPLC and MS methods used in release testing. Without such a reference standard, distinguishing between the active drug and its close analogs becomes speculative, posing significant risks during regulatory audits and batch release certifications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of Sugammadex Sodium involves a Vilsmeier-Hack reaction using triphenylphosphine and iodine to activate the cyclodextrin hydroxyl groups, followed by substitution with 3-mercaptopropionic acid. 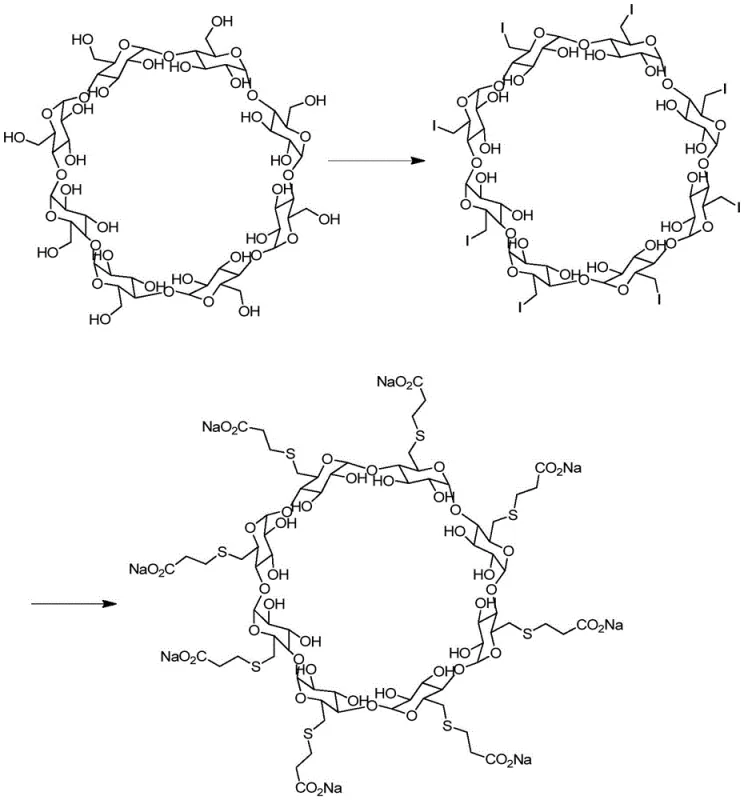
However, conventional approaches to generating the associated impurities for quality control often suffer from poor reproducibility and lack of specificity. In many legacy processes, the formation of phosphine-containing by-products is uncontrolled, occurring randomly depending on slight variations in reagent quality or reaction stoichiometry. This randomness makes it nearly impossible to isolate sufficient quantities of a single, pure impurity species for use as a reference standard. Furthermore, older methods frequently rely on harsh reaction conditions that can degrade the sensitive cyclodextrin backbone, leading to a complex mixture of degradation products rather than the target analog. This complexity complicates the purification process, often requiring extensive chromatographic separation that drastically reduces overall recovery and increases the cost of goods. Consequently, many manufacturers struggle to secure a consistent supply of high-purity impurity standards, leading to bottlenecks in their quality control laboratories and delays in batch release.
The Novel Approach
The methodology outlined in patent CN110818817A offers a decisive solution by deliberately synthesizing the impurity through a controlled, step-wise pathway. Instead of relying on random side reactions, this approach utilizes diphenylphosphine oxide methanol as a specific reagent to introduce the phosphine oxide moiety onto the cyclodextrin scaffold. This deliberate strategy ensures that the resulting product is structurally identical to the process-related impurity found in the main API synthesis, providing a true match for analytical comparison. The process is designed to be operationally convenient, utilizing common organic solvents such as N,N-dimethylformamide (DMF) or dimethyl sulfoxide (DMSO), which are readily available in most pharmaceutical manufacturing facilities. By controlling the stoichiometry and reaction parameters, the method minimizes the formation of unrelated by-products, thereby simplifying the downstream purification workload. This targeted synthesis not only improves the purity of the final reference standard but also enhances the stability of the reaction, making it scalable for the production of larger batches required by high-volume API manufacturers.
Mechanistic Insights into Nucleophilic Substitution on Iodinated Cyclodextrin
The core chemical transformation driving this synthesis is a nucleophilic substitution reaction on a highly functionalized cyclodextrin derivative. The process begins with the activation of 3-mercaptopropionic acid and diphenylphosphine oxide methanol using sodium hydride (NaH) as a strong base. 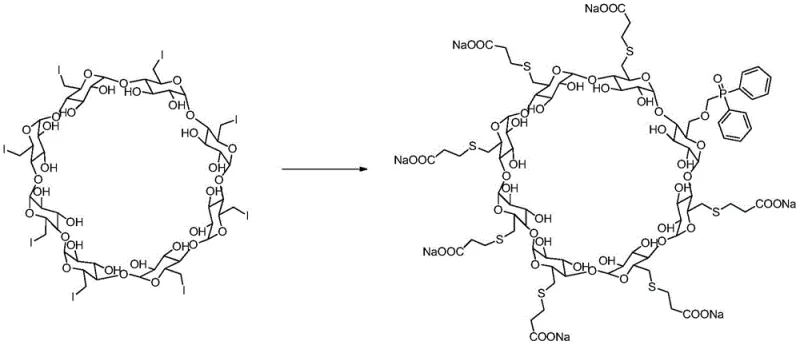
This activation step generates the corresponding thiolate and phosphinate anions, which act as potent nucleophiles. The substrate, 6-per-deoxy-6-per-iodo-γ-cyclodextrin, possesses excellent leaving groups (iodine atoms) at the primary hydroxyl positions, making it highly susceptible to nucleophilic attack. The reaction mechanism proceeds via an SN2 pathway, where the nucleophile attacks the carbon bearing the iodine atom, displacing the iodide ion and forming a stable carbon-sulfur or carbon-phosphorus bond. The patent specifies maintaining the temperature at ≤5°C during the addition of sodium hydride to control the exothermic nature of the deprotonation and prevent premature side reactions. Once the nucleophiles are generated, the temperature is raised to 55-60°C to provide the necessary activation energy for the substitution on the sterically hindered cyclodextrin ring. This precise thermal profile is crucial for balancing reaction rate with selectivity, ensuring that the substitution occurs primarily at the desired positions without causing elimination reactions or backbone degradation.
Controlling the impurity profile during this synthesis is achieved through rigorous management of reagent ratios and post-processing conditions. The patent recommends a molar ratio of 6-per-deoxy-6-per-iodo-γ-cyclodextrin to 3-mercaptopropionic acid and diphenylphosphine oxide methanol ranging from 1:6~10:1~3. This excess of nucleophiles drives the reaction to completion, ensuring that the limited quantity of the expensive iodinated cyclodextrin is fully utilized. Following the reaction, the workup procedure involves cooling the mixture to 18-20°C and slowly adding water and methanol to precipitate the crude product. This anti-solvent crystallization technique effectively separates the organic product from inorganic salts and residual solvents. The subsequent washing with methanol further removes soluble impurities, yielding a crude solid that is already enriched in the target compound. Final purification, likely via preparative chromatography or recrystallization, yields the single compound of Formula I with high purity, suitable for use as a certified reference material in high-performance liquid chromatography (HPLC) and mass spectrometry (MS) assays.
How to Synthesize Sugammadex Sodium Impurity Efficiently
Implementing this synthesis route requires adherence to strict operational parameters to ensure safety and reproducibility. The use of sodium hydride necessitates an inert atmosphere, typically nitrogen or argon, to prevent moisture ingress which could lead to hydrogen gas evolution and potential safety hazards. The detailed standardized synthesis steps involve precise weighing of reagents, controlled addition rates, and specific temperature ramps that must be monitored continuously. For R&D teams looking to replicate this reference standard, it is essential to utilize high-purity solvents and dry glassware to maintain the integrity of the anionic intermediates. The post-reaction handling also demands care, particularly during the quenching phase where water is added to the reaction mixture.
- Dissolve 3-mercaptopropionic acid and diphenylphosphine oxide methanol in an organic solvent like DMF or DMSO under inert gas protection.
- Add sodium hydride at controlled low temperatures (≤5°C) to activate the nucleophile, followed by the addition of 6-per-deoxy-6-per-iodo-γ-cyclodextrin.
- Maintain reaction temperature between 55-60°C for 16-20 hours, followed by quenching, filtration, and purification to isolate the target impurity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this optimized synthesis method translates into tangible strategic benefits beyond mere technical compliance. The ability to internally generate or reliably source this critical impurity standard mitigates the risk of supply disruptions that often plague the specialty chemicals market. By reducing reliance on external vendors who may have long lead times or inconsistent quality, pharmaceutical companies can maintain greater control over their quality control timelines. This autonomy is particularly valuable during regulatory inspections where immediate access to validated reference standards is often required to verify test results. Furthermore, the streamlined nature of the reaction reduces the consumption of exotic reagents, aligning with broader initiatives to simplify the supply chain and reduce procurement complexity.
- Cost Reduction in Manufacturing: The elimination of complex, multi-step purification sequences traditionally required to isolate trace impurities leads to significant operational savings. By designing a route that selectively produces the target impurity, the need for extensive preparative HPLC runs is minimized, reducing solvent consumption and labor costs. Additionally, the use of common industrial solvents like DMF and DMSO avoids the premium pricing associated with specialized chromatographic grades. The high stability of the reaction also means fewer failed batches and less waste disposal, contributing to a leaner cost structure for quality control operations. These efficiencies accumulate over time, providing a substantial reduction in the total cost of ownership for analytical reference materials.
- Enhanced Supply Chain Reliability: Sourcing high-purity impurity standards from the open market can be unpredictable, with suppliers often facing raw material shortages or production bottlenecks. By utilizing a synthesis method based on readily available starting materials such as 3-mercaptopropionic acid and cyclodextrin derivatives, manufacturers can secure a more resilient supply chain. The robustness of the reaction conditions allows for production in multiple facilities, diversifying the supply base and reducing the risk of single-source dependency. This reliability ensures that quality control laboratories never face stock-outs of critical standards, preventing delays in batch release and shipment of the final API to customers. Consistent availability supports continuous manufacturing schedules and strengthens relationships with downstream partners.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that can be safely transferred from laboratory glassware to pilot plant reactors. The moderate temperature range and atmospheric pressure operation simplify equipment requirements, allowing for production in standard multipurpose facilities without the need for specialized high-pressure or cryogenic infrastructure. From an environmental perspective, the efficient use of reagents and the ability to recover solvents during the workup phase contribute to a reduced environmental footprint. The generation of less hazardous waste compared to traditional isolation methods aligns with green chemistry principles and helps companies meet increasingly stringent environmental regulations. This sustainability aspect is becoming a key differentiator in vendor selection processes for major pharmaceutical corporations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this specific impurity standard. Understanding these details is crucial for quality assurance teams and technical procurement officers who are responsible for validating analytical methods and sourcing materials. The answers are derived directly from the technical specifications and beneficial effects described in the patent literature, ensuring accuracy and relevance to real-world manufacturing scenarios.
Q: Why is the diphenyl phosphine oxide derivative impurity critical for Sugammadex Sodium?
A: This specific impurity arises from the use of triphenylphosphine in the main API synthesis. Regulatory bodies require strict identification and quantification limits for such toxicologically non-characteristic impurities to ensure patient safety.
Q: What are the key reaction conditions for synthesizing this impurity standard?
A: The process requires precise temperature control, starting with sodium hydride addition at ≤5°C to prevent side reactions, followed by a sustained reaction phase at 55-60°C to ensure complete conversion of the iodinated cyclodextrin intermediate.
Q: How does this synthesis method improve supply chain reliability?
A: By utilizing stable, commercially available starting materials and mild reaction conditions, the method reduces dependency on exotic reagents, thereby minimizing procurement risks and ensuring consistent availability of critical reference standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sugammadex Sodium Impurity Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your analytical data is only as good as the reference standards you rely upon. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that even complex molecules like cyclodextrin derivatives are handled with the utmost precision. We are committed to delivering materials that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of hard-to-find impurities or large-scale supply of key intermediates, our infrastructure is designed to support your regulatory and commercial goals without compromise.
We invite you to collaborate with us to optimize your supply chain and enhance your quality control capabilities. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific production volumes and purity requirements. Contact us today to request specific COA data and route feasibility assessments for your next project. Let us help you secure a competitive advantage through superior chemical solutions and unwavering supply reliability.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →