Advanced Three-Step Synthesis of Cytosine Nucleoside for Scalable Antiviral API Manufacturing
The pharmaceutical industry continuously seeks robust and economically viable pathways for the production of essential nucleoside intermediates, particularly for antiviral and antitumor applications. Patent CN112480197A introduces a groundbreaking methodology for the synthesis of Cytosine Nucleoside (CAS: 65-46-3), a critical building block for medications such as arabinosyl cytosine and citicoline. This innovation departs from traditional reliance on expensive natural nucleosides or complex protected bases, instead utilizing cyanoacetaldehyde urea acetal as a cost-efficient starting material. The disclosed process streamlines production into a continuous three-step sequence that integrates silylation, Lewis acid-catalyzed glycosylation, and a final one-pot cyclization-deprotection stage. By addressing the longstanding challenges of isomer separation and harsh reaction conditions found in legacy methods, this technology offers a compelling solution for reliable pharmaceutical intermediate supplier networks aiming to optimize their manufacturing portfolios for next-generation therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Cytosine Nucleoside has been plagued by significant operational and economic inefficiencies that hinder large-scale commercial viability. Early methods, such as those proposed by Nishimura, relied on the condensation of silyl-protected N4-acetyl cytosine with chlorotriphenylformyl ribose, resulting in complex mixtures of alpha and beta isomers that required difficult and yield-reducing chromatographic separations. Alternative approaches utilizing uridine as a starting material necessitated extreme conditions, including high-pressure ammonolysis at 165°C and 25 atmospheres, which pose severe safety risks and equipment costs for industrial plants. Furthermore, routes employing fully protected cytosine derivatives often suffer from the hygroscopic nature of intermediates and the high cost of reagents like isobutyric anhydride or tribenzoyl ribose. These conventional pathways are characterized by low overall yields, multi-step isolation procedures, and a heavy dependence on scarce or expensive biochemical reagents, making cost reduction in pharmaceutical intermediate manufacturing nearly impossible without process innovation.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach detailed in the patent leverages a strategic redesign of the synthetic route to maximize efficiency and minimize waste. By initiating the synthesis with cyanoacetaldehyde urea acetal, the process bypasses the need for pre-formed cytosine rings, effectively reducing raw material costs substantially. The methodology employs a telescoped strategy where the silylated intermediate is condensed directly with tetraacetyl ribose under Lewis acid catalysis, followed immediately by a base-mediated ring closure. This integration allows for the elimination of intermediate isolation steps between the glycosylation and cyclization phases. The final transformation utilizes sodium alkoxide to simultaneously effect cyclization and deprotection of the acetyl groups, yielding the target nucleoside in high purity through simple recrystallization. This streamlined workflow not only simplifies the operational complexity but also enhances the stability of yields during scale-up, providing a distinct competitive advantage for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Lewis Acid Catalyzed Glycosylation and Cyclization
The core of this synthetic strategy lies in the precise manipulation of nitrogen nucleophilicity and glycosidic bond formation through silylation and Lewis acid activation. In the initial phase, cyanoacetaldehyde urea acetal undergoes silylation using reagents such as hexamethyldisilazane (HMDS) or trimethylchlorosilane (TMCS). This step is crucial as it protects the exocyclic amine and activates the urea moiety, increasing its solubility in organic solvents and enhancing its nucleophilic character for the subsequent attack on the sugar anomeric center. The silyl ether product serves as a stable yet reactive species that prevents unwanted side reactions during the glycosylation event. Following this, the introduction of a potent Lewis acid catalyst, such as stannic chloride (SnCl4) or titanium tetrachloride (TiCl4), activates the tetraacetyl ribose by coordinating with the anomeric acetate group. This coordination facilitates the departure of the acetate leaving group, generating a highly reactive oxocarbenium ion intermediate that is rapidly attacked by the silylated nitrogen nucleophile to form the N-glycosidic bond with high stereoselectivity.
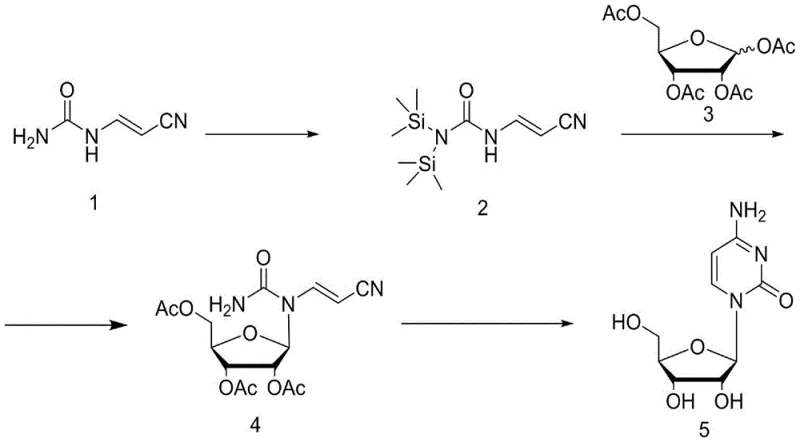
Following the formation of the condensed intermediate, the process transitions to a sophisticated base-mediated cyclization and deprotection mechanism. The addition of sodium alkoxide, such as sodium methoxide or sodium ethoxide, in an alcoholic solvent serves a dual purpose that is critical for the efficiency of this route. Firstly, the alkoxide acts as a base to deprotect the acetyl groups on the ribose sugar, regenerating the free hydroxyl groups necessary for the biological activity of the final nucleoside. Simultaneously, the basic conditions promote an intramolecular cyclization where the amino group attacks the nitrile functionality of the side chain, closing the pyrimidine ring to form the cytosine base structure in situ. This tandem reaction eliminates the need for separate hydrolysis and ring-closing steps, thereby reducing solvent consumption and processing time. The result is a highly pure product that precipitates directly from the reaction mixture, demonstrating superior impurity control mechanisms compared to traditional multi-step syntheses that often generate persistent byproducts requiring extensive purification.
How to Synthesize Cytosine Nucleoside Efficiently
The execution of this synthesis requires careful attention to reaction parameters to ensure optimal conversion and product quality. The process begins with the silylation of the urea acetal precursor in a solvent such as dichloroethane or dichloromethane, typically under reflux conditions to drive the equilibrium towards the silylated product. Once the silylation is complete, the reaction mixture is cooled, and tetraacetyl ribose is introduced along with the Lewis acid catalyst at controlled low temperatures to manage the exotherm and ensure regioselectivity. After the condensation is verified by TLC, the mixture is quenched and worked up to isolate the condensed intermediate, which is then subjected to the final cyclization step using sodium alkoxide in methanol or ethanol under reflux. For a comprehensive guide on the specific molar ratios, temperature profiles, and workup procedures validated in the patent examples, please refer to the standardized protocol below.
- Silylation of cyanoacetaldehyde urea acetal using hexamethyldisilazane (HMDS) or trimethylchlorosilane (TMCS) to form the silyl ether intermediate.
- Condensation of the silyl ether product with tetraacetyl ribose under Lewis acid catalysis (e.g., SnCl4, TiCl4) at low temperature followed by warming.
- One-pot cyclization and deprotection using sodium alkoxide (e.g., sodium methoxide) in alcohol solvent under reflux to yield pure Cytosine Nucleoside.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route presents a transformative opportunity to enhance margin structures and secure supply continuity for critical antiviral ingredients. The shift from expensive natural nucleosides like uridine to synthetic precursors like cyanoacetaldehyde urea acetal fundamentally alters the cost basis of the final API intermediate. By utilizing commodity chemicals that are readily available in the bulk market, manufacturers can insulate themselves from the volatility associated with bio-fermented starting materials. Furthermore, the telescoped nature of the process reduces the number of unit operations, which directly correlates to lower labor costs, reduced energy consumption, and decreased solvent usage. This operational efficiency translates into significant cost savings in pharmaceutical intermediate manufacturing without compromising the stringent quality standards required for regulatory compliance.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the substitution of high-cost starting materials with affordable synthetic alternatives and the elimination of chromatographic purification. Traditional methods often require expensive protected sugars and bases, alongside costly silica gel columns for isomer separation, which generate substantial solid waste. In this new route, the final product is purified via recrystallization from ethanol, a low-cost and easily recoverable solvent. Additionally, the one-pot cyclization and deprotection step consolidates two distinct chemical transformations into a single vessel operation, drastically reducing reactor occupancy time and utility costs. These factors combine to create a leaner manufacturing profile that supports aggressive pricing strategies while maintaining healthy profit margins.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the reliance on stable, non-perishable chemical feedstocks rather than sensitive biochemical reagents. The starting materials, including tetraacetyl ribose and cyanoacetaldehyde urea acetal, are produced by established chemical suppliers with robust global distribution networks, minimizing the risk of stockouts or delivery delays. The process tolerance is also improved, as the reaction conditions are less sensitive to moisture and oxygen compared to high-pressure ammonolysis methods, allowing for more flexible scheduling and reduced batch failure rates. This reliability ensures reducing lead time for high-purity pharmaceutical intermediates, enabling downstream API manufacturers to maintain consistent production schedules and meet market demand fluctuations effectively.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this methodology aligns perfectly with modern green chemistry principles and industrial safety standards. The avoidance of high-pressure reactors and extreme temperatures reduces the capital expenditure required for plant infrastructure and lowers the safety risk profile for operators. The reduction in solvent volume due to the telescoped steps and the use of recyclable alcohols for crystallization minimizes the generation of hazardous waste streams. This simplified waste profile facilitates easier compliance with increasingly strict environmental regulations regarding effluent discharge. Consequently, the process is inherently easier to scale from pilot plant quantities to multi-ton commercial production, offering a clear pathway for rapid capacity expansion to support growing global demand for nucleoside-based therapeutics.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical advantages of this route over incumbent methods. Understanding these details is essential for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the long-term value proposition of this manufacturing strategy.
Q: Why is cyanoacetaldehyde urea acetal preferred over uridine for Cytosine Nucleoside synthesis?
A: Cyanoacetaldehyde urea acetal is significantly more cost-effective and readily available compared to uridine or protected cytosine derivatives. Traditional methods using uridine require harsh high-pressure ammonolysis conditions, whereas this novel route utilizes mild Lewis acid catalysis and standard reflux conditions, greatly simplifying industrial scale-up.
Q: How does this process achieve high purity without column chromatography?
A: The process is designed as a telescoped three-step sequence where intermediates are carried forward or simply quenched and extracted. The final cyclization and deprotection step precipitates the product directly from the reaction mixture. Final purification is achieved solely through recrystallization from ethanol, eliminating the need for expensive and time-consuming silica gel chromatography.
Q: What are the critical reaction conditions for the glycosylation step?
A: The glycosylation step requires strict temperature control, typically initiating at -5°C to 0°C during the addition of the Lewis acid catalyst (such as stannic chloride or titanium tetrachloride) to prevent side reactions. The reaction is then warmed to approximately 35°C to drive the condensation to completion, ensuring high conversion rates before proceeding to the final cyclization.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cytosine Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes in the competitive landscape of antiviral drug development. Our team of expert chemists has thoroughly analyzed the potential of this patented three-step process and is fully prepared to assist partners in translating this laboratory innovation into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop to plant floor is seamless and compliant with cGMP standards. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of Cytosine Nucleoside delivered meets the highest international quality benchmarks required for API synthesis.
We invite pharmaceutical companies and contract manufacturers to collaborate with us to leverage this advanced technology for their supply chains. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and logistical needs. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in nucleoside chemistry can drive value and security for your critical drug programs.