Advanced Synthetic Route for Remdesivir Key Intermediates via Novel Pyrrolo-Triazine Derivatives
Introduction to Next-Generation Remdesivir Synthesis
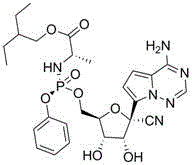
The global demand for effective antiviral therapeutics has placed immense pressure on the pharmaceutical supply chain to deliver high-purity active pharmaceutical ingredients (APIs) with unprecedented speed and reliability. Patent CN111233869B introduces a groundbreaking methodology for synthesizing key intermediates of Remdesivir, addressing critical bottlenecks that have historically plagued large-scale manufacturing. This technology centers on the preparation of a novel compound II, which serves as a superior precursor to the final drug substance by utilizing a strategic masking group approach on the pyrrolo[2,1-f][1,2,4]triazine core. Unlike traditional methods that struggle with the reactivity of free amino groups, this innovation employs halogen or methylthio substituents to enhance reaction selectivity and overall process efficiency.
For procurement leaders and R&D directors seeking a reliable pharmaceutical intermediate supplier, understanding the structural nuances of this new pathway is essential. The ability to bypass complex protection strategies translates directly into reduced operational complexity and lower cost of goods sold (COGS). As we delve into the technical specifics, it becomes clear that this patent represents not just a chemical improvement, but a strategic asset for securing the supply chain of vital antiviral medications. The following analysis details how this novel chemistry overcomes the inherent limitations of previous generations of synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Remdesivir intermediates has relied heavily on routes developed by originator companies, which often involve starting materials containing free amino groups on the heterocyclic ring. As illustrated in the prior art, these conventional pathways necessitate the use of 4-amino-7-bromo or 4-amino-7-iodo-pyrrolo[2,1-f][1,2,4]triazine derivatives. The presence of the primary amine functionality introduces significant chemical challenges, particularly its nucleophilicity and basicity, which interfere with subsequent metal-halogen exchange reactions required for coupling with the sugar moiety.
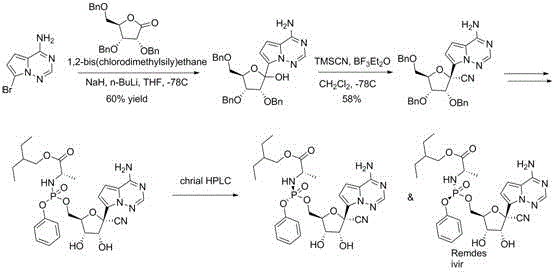
To mitigate these issues, existing processes mandate the use of silane protecting groups to mask the amine prior to lithiation. This additional step not only increases the number of unit operations but also introduces variability in yield and purity due to the difficulty of achieving complete protection and the sensitivity of organolithium reagents at cryogenic temperatures. Furthermore, the lithiation step itself is prone to side reactions and often results in inconsistent yields, reported as low as 40% in some optimized variations. These inherent structural defects in the starting material create a fragile process window that is ill-suited for the robust demands of commercial-scale API manufacturing.
The Novel Approach
The innovative strategy disclosed in CN111233869B fundamentally reimagines the synthetic entry point by replacing the problematic amino group with a chemically inert masking group, denoted as X, which can be fluorine, chlorine, bromine, iodine, or a methylthio group. This subtle yet profound modification eliminates the need for amino protection entirely, thereby streamlining the synthesis and removing a major source of impurity formation. The new compound II, featuring this masked triazine core coupled to the ribose sugar, demonstrates superior stability and reactivity profiles during the critical carbon-carbon bond-forming steps.
By utilizing this masked intermediate, the process achieves significantly higher reaction selectivity and enables batch preparation with improved consistency. The masking group X remains stable during the Grignard addition and cyanation steps, only to be converted to the requisite amino group in a final, high-yielding amination step using ammonia. This late-stage functionalization strategy ensures that the sensitive coupling reactions proceed without interference from basic nitrogen species, resulting in a cleaner reaction profile and facilitating easier purification. For organizations focused on cost reduction in pharmaceutical intermediate manufacturing, this elimination of protection-deprotection cycles offers a direct path to enhanced economic efficiency.
Mechanistic Insights into Grignard Coupling and Cyanation
The core of this technological advancement lies in the precise execution of the Grignard addition and subsequent cyanation, facilitated by the unique electronic properties of the masked triazine ring. In the first critical transformation, the 4-X-7-halo-pyrrolo[2,1-f][1,2,4]triazine undergoes magnesium-halogen exchange using turbo-Grignard reagents such as iPrMgCl·LiCl. The presence of the electron-withdrawing or neutral masking group at the 4-position stabilizes the heterocyclic ring against nucleophilic attack by the Grignard reagent, ensuring that metalation occurs exclusively at the 7-position. This regioselectivity is paramount for generating the correct organomagnesium species required for coupling with the ribolactone derivative.
Following the formation of the glycosidic bond, the resulting hemiaminal intermediate must be converted into the quaternary nitrile center, a defining feature of the Remdesivir scaffold. This is achieved through a Lewis acid-catalyzed cyanation using trimethylsilyl cyanide (TMSCN) and a catalyst such as trimethylsilyl trifluoromethanesulfonate (TMSOTf). The mechanism involves the activation of the hydroxyl group by the Lewis acid, generating an oxocarbenium ion-like intermediate that is subsequently trapped by the cyanide nucleophile. The steric and electronic environment provided by the masked triazine ring favors the formation of the desired stereochemistry at the anomeric center, minimizing the formation of diastereomeric impurities that are difficult to separate.
Impurity control is further enhanced by the choice of protecting groups on the ribose sugar, typically benzyl or silyl ethers, which remain orthogonal to the reaction conditions employed. The robustness of the masking group X ensures that no competitive side reactions, such as nucleophilic aromatic substitution or ring opening, occur during the acidic cyanation step. This high level of chemoselectivity translates to a crude product profile that is significantly cleaner than that obtained from amino-protected routes, reducing the burden on downstream purification processes like chromatography or crystallization. For R&D teams, this implies a more predictable and controllable process capable of delivering high-purity Remdesivir intermediates consistently.
How to Synthesize Remdesivir Key Intermediate Efficiently
The practical implementation of this synthesis involves a sequence of well-defined chemical transformations that leverage commercially available reagents and standard processing equipment. The process begins with the halogenation of the triazine core, followed by the crucial Grignard coupling with the sugar lactone, and concludes with the cyanation to install the nitrile group. Each step has been optimized to balance reaction rate with selectivity, ensuring that the process can be transferred from the laboratory to pilot and production scales with minimal risk. Detailed standard operating procedures for temperature control, reagent addition rates, and workup protocols are essential to replicate the high yields reported in the patent examples.
- Halogenation of 4-X-pyrrolo[2,1-f][1,2,4]triazine using NIS or brominating agents to activate the 7-position.
- Grignard addition using iPrMgCl·LiCl to couple the activated triazine with protected ribolactone at low temperature (-20°C).
- Cyanation of the resulting hemiaminal using TMSCN and TMSOTf to install the critical quaternary nitrile center.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this novel synthetic route offers transformative benefits for procurement managers and supply chain directors tasked with securing reliable sources of complex antiviral intermediates. The primary advantage stems from the drastic simplification of the synthetic sequence, which directly correlates to reduced manufacturing costs and shorter production cycles. By eliminating the need for amino protection and the associated reagents, the process reduces raw material consumption and waste generation, aligning with both economic and environmental sustainability goals. This efficiency gain is critical for maintaining competitiveness in the volatile market for generic antiviral APIs.
- Cost Reduction in Manufacturing: The removal of the amino protection-deprotection sequence represents a significant saving in both reagent costs and processing time. Traditional routes require expensive silylating agents and additional purification steps to remove protecting group byproducts, all of which add to the overall cost of goods. Furthermore, the higher yields achieved in the Grignard and cyanation steps (often exceeding 70-80% compared to 40% in prior art) mean that less starting material is required to produce the same amount of final product. This improvement in atom economy and process mass intensity (PMI) drives down the unit cost, allowing for more aggressive pricing strategies while maintaining healthy margins.
- Enhanced Supply Chain Reliability: The reagents utilized in this novel pathway, such as iPrMgCl·LiCl and TMSCN, are commodity chemicals available from multiple global suppliers, reducing the risk of single-source dependency. In contrast, specialized amino-protected heterocycles often require custom synthesis with long lead times, creating potential bottlenecks in the supply chain. The robustness of the new chemistry also means that production schedules are less likely to be disrupted by batch failures or quality deviations. This reliability is essential for meeting the rigorous delivery commitments expected by top-tier pharmaceutical clients and ensures continuity of supply for critical medications.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that are amenable to large-scale reactor operations without requiring exotic equipment. The avoidance of cryogenic lithiation (-78°C) in favor of milder Grignard conditions (-20°C to 0°C) reduces energy consumption and simplifies thermal management in production plants. Additionally, the cleaner reaction profile results in less hazardous waste and simpler effluent treatment, facilitating compliance with increasingly stringent environmental regulations. This ease of commercial scale-up of complex nucleoside analogs positions manufacturers to rapidly respond to surges in demand without compromising on quality or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear understanding of the value proposition for potential partners. Understanding these details is crucial for making informed decisions about technology licensing or procurement strategies.
Q: Why is the amino group replaced by a halogen or methylthio group in this synthesis?
A: Replacing the amino group with a masking group like chlorine or methylthio prevents unwanted side reactions during the Grignard coupling and eliminates the need for cumbersome protection-deprotection sequences, significantly improving yield and purity.
Q: What are the critical reaction conditions for the cyanation step?
A: The cyanation requires a Lewis acid catalyst such as TMSOTf and a cyanating agent like TMSCN in dichloromethane at low temperatures (-20°C) to ensure high stereoselectivity and prevent decomposition of the sensitive glycosidic bond.
Q: How does this route improve scalability compared to prior art?
A: By avoiding cryogenic lithium-halogen exchange and unstable amino-protected intermediates, this route utilizes robust Grignard reagents and achieves consistent yields above 70%, making it far more suitable for multi-kilogram commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Remdesivir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient and scalable synthetic routes for high-value antiviral intermediates. Our team of expert process chemists has extensively evaluated the methodology described in CN111233869B and confirmed its potential for industrial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to full-scale manufacturing is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required for regulatory filings, guaranteeing that every batch of Remdesivir intermediate meets the highest global standards.
We invite pharmaceutical companies and generic manufacturers to collaborate with us to optimize their supply chains using this advanced technology. By leveraging our expertise in heterocyclic chemistry and nucleoside synthesis, we can help you achieve significant Customized Cost-Saving Analysis tailored to your specific production volumes. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us partner with you to secure a resilient and cost-effective supply of critical antiviral intermediates for the global market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →