Scalable Production of Structurally Diverse Beta-Amino Acids via Novel C-H Activation
Scalable Production of Structurally Diverse Beta-Amino Acids via Novel C-H Activation
The pharmaceutical industry continuously seeks robust and versatile synthetic routes for high-value building blocks, particularly beta-amino acids which serve as critical precursors for antibiotics like penicillins and complex anticancer agents such as Taxol. Patent CN107793351B introduces a groundbreaking methodology that leverages direct carbon-hydrogen bond amination to construct these valuable scaffolds with unprecedented efficiency. Unlike conventional approaches that often struggle with regioselectivity or require pre-functionalized starting materials, this invention utilizes a palladium-catalyzed system to directly install nitrogen functionality onto simple amide substrates. The process begins with the reaction of specific amides and azodicarboxylates to form a key hydrazine intermediate, followed by alkylation and subsequent deprotection to yield the target beta-amino acid. This technological advancement represents a significant leap forward in organic synthesis, offering a pathway to structurally diverse molecules that were previously difficult or expensive to access through traditional means.
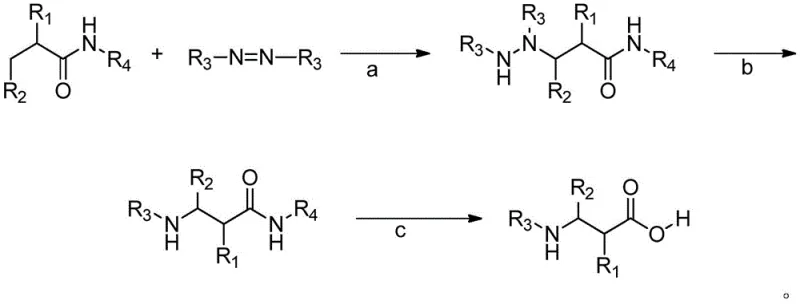
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of beta-amino acids has relied heavily on methods that impose significant constraints on both safety and structural variety. Traditional routes frequently employ toxic reagents such as ethyl cyanoacetate or involve multi-step sequences with poor atom economy, leading to substantial waste generation and higher processing costs. For instance, earlier patents describe one-step hydrogenation methods that achieve yields around 70 percent but fail to introduce substituents at both the alpha and beta positions simultaneously, resulting in products with limited structural diversity. Furthermore, methods utilizing malonic acid derivatives or benzylamine often require harsh conditions or expensive noble metal catalysts like palladium-carbon for hydrogenolysis, which complicates the purification process and increases the risk of metal contamination in the final active pharmaceutical ingredient. These legacy techniques restrict the ability of medicinal chemists to rapidly explore chemical space, thereby slowing down the drug discovery pipeline and increasing the overall cost of goods for potential therapies.
The Novel Approach
In stark contrast, the methodology disclosed in CN107793351B offers a streamlined and highly adaptable solution by employing direct C-H bond functionalization. This innovative route bypasses the need for pre-activated halides or toxic cyanide sources, instead utilizing readily available amides and azodicarboxylates as primary feedstocks. The core transformation involves a palladium-catalyzed amination that selectively targets the beta-C-H bond, guided by a specific amide directing group such as 8-aminoquinoline or a pyridyl-ethylamine derivative. This strategic use of a directing group ensures high regioselectivity, allowing for the precise installation of the nitrogen moiety even in the presence of other reactive sites. Following the initial amination, the intermediate undergoes a smooth alkylation with methyl bromoacetate and a final acidic hydrolysis step to reveal the free amino acid. This sequence not only simplifies the operational workflow but also dramatically expands the scope of accessible substrates, enabling the production of beta-amino acids with varied alkyl, aryl, and heterocyclic substituents that are essential for modern drug design.
Mechanistic Insights into Pd-Catalyzed C-H Amination
The success of this synthesis hinges on the sophisticated interplay between the palladium catalyst and the directing group attached to the amide nitrogen. Mechanistically, the reaction initiates with the coordination of the palladium species to the nitrogen atom of the directing group, which brings the metal center into close proximity with the target beta-carbon-hydrogen bond. This proximity facilitates the activation of the inert C-H bond through a cyclometallation process, forming a stable palladacycle intermediate. Subsequently, the azodicarboxylate acts as an electrophilic nitrogen source, inserting into the palladium-carbon bond to generate the hydrazine-linked Intermediate I. This step is critical as it determines the overall efficiency and selectivity of the transformation, with the choice of ligand and solvent playing a pivotal role in stabilizing the active catalytic species. The use of oxygen protection during this stage prevents the oxidation of sensitive intermediates, ensuring that the reaction proceeds with high fidelity and minimal formation of oxidative byproducts.
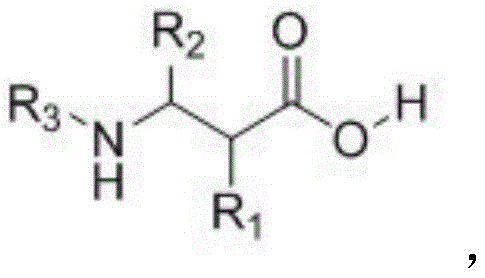
Following the formation of the hydrazine intermediate, the process moves to an alkylation step where methyl bromoacetate reacts with the nitrogen center to extend the carbon chain, effectively setting up the beta-amino acid skeleton. The final step involves the removal of the amide protecting group under acidic conditions, typically using hydrochloric acid at elevated temperatures. This deprotection strategy is designed to be robust enough to cleave the strong amide bond without degrading the sensitive ester or amino functionalities elsewhere in the molecule. From an impurity control perspective, the specificity of the C-H activation step significantly reduces the formation of regioisomers, which are common contaminants in non-directed functionalization reactions. Furthermore, the use of well-defined palladium catalysts allows for better control over metal residues, facilitating easier downstream purification to meet the stringent purity specifications required for pharmaceutical intermediates. The ability to tune the R1 and R2 groups on the starting amide provides a powerful handle for optimizing the physicochemical properties of the final product, ensuring that the synthesis can be adapted to meet the specific needs of different therapeutic programs.
How to Synthesize Beta-Amino Acid Efficiently
The practical implementation of this synthesis route involves a carefully orchestrated three-step sequence that balances reaction efficiency with operational simplicity. The process begins with the preparation of the appropriate amide substrate containing the necessary directing group, which is then subjected to the palladium-catalyzed amination conditions in a suitable solvent such as 2-methyl-2-butanol or toluene. Once the first intermediate is isolated, it is immediately engaged in the alkylation reaction with methyl bromoacetate using cesium carbonate as a base in acetonitrile, a step that proceeds rapidly at moderate temperatures. The final hydrolysis step requires careful control of temperature and acid concentration to ensure complete deprotection while minimizing side reactions. Detailed standardized operating procedures for each of these stages, including specific molar ratios, temperature profiles, and workup protocols, are essential for achieving consistent results across different batches and scales of production.
- Perform direct carbon-hydrogen bond amination of an amide substrate with an azodicarboxylate using a palladium catalyst under oxygen protection to form Intermediate I.
- React Intermediate I with methyl bromoacetate in the presence of cesium carbonate and acetonitrile to synthesize Intermediate II through alkylation.
- Treat Intermediate II with hydrochloric acid at elevated temperatures to remove the amide protecting group and yield the final beta-amino acid product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis method offers compelling advantages rooted in safety, flexibility, and cost efficiency. By eliminating the reliance on hazardous cyanide reagents and high-pressure hydrogenation equipment, the process significantly lowers the barrier for entry regarding facility safety compliance and insurance costs. The modular nature of the synthesis allows manufacturers to utilize a broad range of commercially available amides and azo compounds, reducing the risk of supply chain disruptions associated with specialized or single-source raw materials. Moreover, the mild reaction conditions translate to lower energy consumption and reduced wear on reactor equipment, contributing to a more sustainable and economically viable manufacturing operation that aligns with modern green chemistry principles.
- Cost Reduction in Manufacturing: The elimination of toxic cyanide sources and the avoidance of expensive noble metal catalysts for hydrogenolysis directly contribute to a leaner cost structure. By utilizing standard palladium catalysts in low loadings and avoiding complex purification steps associated with heavy metal removal, the overall processing costs are significantly optimized. Additionally, the high atom economy of the direct C-H functionalization step minimizes waste disposal fees, further enhancing the economic attractiveness of this route for large-scale production.
- Enhanced Supply Chain Reliability: The versatility of the substrate scope means that manufacturers are not locked into a single supply line for specialized precursors. Since the method accommodates various alkyl, aryl, and heterocyclic groups, procurement teams can source alternative starting materials more easily if a specific supplier faces shortages. This flexibility ensures continuous production capability and reduces the lead time for high-purity pharmaceutical intermediates, safeguarding the manufacturing schedule against external market volatility.
- Scalability and Environmental Compliance: The use of common organic solvents like acetonitrile and toluene, combined with atmospheric pressure reactions, simplifies the engineering requirements for scale-up. This ease of translation from bench to plant reduces the time and capital investment needed for process validation. Furthermore, the reduced generation of hazardous waste and the absence of highly toxic reagents streamline environmental permitting and compliance reporting, making it a preferred choice for facilities aiming to maintain rigorous environmental standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this beta-amino acid synthesis technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing a clear understanding of the method's capabilities and limitations for potential partners and licensees.
Q: How does this Pd-catalyzed method improve safety compared to traditional cyanide-based routes?
A: Traditional methods often rely on toxic cyanide reagents or harsh hydrogenation conditions. This patent utilizes direct C-H amination with azodicarboxylates, eliminating the need for hazardous cyanide sources and significantly reducing environmental and operational safety risks in the manufacturing facility.
Q: What structural diversity can be achieved with this synthesis platform?
A: The method supports a wide range of substituents at both the alpha and beta positions, including alkyl, branched alkyl, cycloalkyl, aromatic, and heterocyclic groups. This flexibility allows for the rapid generation of diverse libraries of beta-amino acid intermediates essential for optimizing drug candidates.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the patent explicitly highlights mild reaction conditions and good reproducibility as key features for industrial application. The use of standard solvents like acetonitrile and toluene, along with commercially available palladium catalysts, facilitates straightforward scale-up from laboratory to multi-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Beta-Amino Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced C-H activation technologies in accelerating the development of next-generation therapeutics. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory methods like the one described in CN107793351B can be seamlessly transitioned to industrial reality. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of beta-amino acid intermediate delivered meets the highest quality standards required by global regulatory bodies.
We invite you to collaborate with us to leverage this cutting-edge synthesis route for your specific drug development programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your project's unique requirements, demonstrating how this method can optimize your budget without compromising quality. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, and let us help you secure a reliable supply of high-quality beta-amino acids for your pipeline.