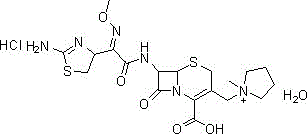
Advanced Green Synthesis of Cefepime Hydrochloride for Commercial Scale-Up
Advanced Green Synthesis of Cefepime Hydrochloride for Commercial Scale-Up
The pharmaceutical industry is constantly seeking more efficient, environmentally compliant, and cost-effective routes for the production of fourth-generation cephalosporins. A pivotal advancement in this domain is detailed in patent CN111171052B, which outlines a superior synthesis method for Cefepime Hydrochloride, a critical broad-spectrum antibiotic. This innovative approach addresses long-standing challenges regarding isomer control and solvent toxicity that have plagued previous manufacturing protocols. By shifting away from hazardous polyhalogenated hydrocarbons towards a greener, Boc-protected pathway, this technology offers a compelling value proposition for global supply chains. The method achieves exceptional purity profiles and yields through a streamlined four-step sequence, starting from readily available 7-amino-3-chloromethyl-3-cephem-4-carboxylic acid diphenylmethyl ester hydrochloride. For R&D directors and procurement specialists alike, understanding the mechanistic nuances of this patent is essential for securing a reliable API intermediate supplier partnership that ensures long-term regulatory compliance and product quality.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Cefepime Hydrochloride has been fraught with significant technical and environmental hurdles, primarily centered around the quaternization step involving N-methylpyrrolidine. Prominent prior art, such as the methods reported by Walker et al. and Naito et al., relied heavily on the use of fluorine-containing polychlorinated hydrocarbons like Freon TF or Carbon Tetrachloride (CCl4) as reaction solvents. While these solvents were effective in precipitating the product to minimize isomerization due to their low solubility properties, they are now strictly prohibited under international environmental protection regulations due to their ozone-depleting potential and high toxicity. Furthermore, these conventional routes often struggled with the formation of delta-2 position isomers, a persistent impurity that complicates downstream purification and reduces overall process efficiency. The reliance on such hazardous materials not only escalates waste disposal costs but also introduces substantial supply chain risks associated with the sourcing of restricted chemicals, making these legacy processes increasingly untenable for modern cost reduction in pharmaceutical manufacturing.
The Novel Approach
In stark contrast to these outdated methodologies, the novel approach disclosed in patent CN111171052B introduces a strategic Boc-protection protocol that fundamentally alters the reaction landscape. By initially protecting the 7-amino group with di-tert-butyl dicarbonate (Boc2O) in tetrahydrofuran, the process effectively masks the nucleophilic site that typically leads to unwanted side reactions. This protection allows the subsequent quaternization with N-methylpyrrolidine to proceed with high regioselectivity in dichloromethane, a solvent that is far more manageable and recyclable than Freon. The resulting inner salt intermediate precipitates cleanly, facilitating easy isolation without the need for complex chromatographic separations. This shift not only eliminates the regulatory burden of handling banned solvents but also simplifies the entire workflow, enabling a seamless transition from laboratory optimization to commercial scale-up of complex antibiotics. The result is a robust, four-step synthesis that delivers high-purity intermediates with significantly reduced environmental impact.
Mechanistic Insights into Boc-Protected Quaternization
The core innovation of this synthesis lies in the precise manipulation of nucleophilicity and steric hindrance during the quaternization phase. In traditional unprotected routes, the free 7-amino group on the cephem nucleus can act as a competing nucleophile or facilitate base-catalyzed isomerization of the double bond within the beta-lactam ring system. By introducing the bulky tert-butoxycarbonyl (Boc) group, the electronic density on the nitrogen atom is reduced, thereby preventing it from interfering with the quaternization of the 3-chloromethyl group by N-methylpyrrolidine. This mechanistic safeguard is crucial for maintaining the integrity of the beta-lactam ring, which is sensitive to alkaline conditions. The reaction proceeds through an SN2 mechanism where the nitrogen of the pyrrolidine ring attacks the chloromethyl carbon, displacing the chloride ion to form the quaternary ammonium salt. Because the amino group is protected, the basicity of the N-methylpyrrolidine does not trigger the rearrangement to the thermodynamically stable but biologically inactive delta-2 isomer, ensuring that the final product retains the desired delta-3 configuration essential for antibacterial activity.
Furthermore, the purification mechanism leverages the unique solubility characteristics of the zwitterionic inner salt formed in the second step. Upon completion of the quaternization reaction in dichloromethane, the removal of the solvent and the addition of ether induce the crystallization of the intermediate. This physical separation technique is highly effective at excluding non-ionic impurities and unreacted starting materials, which remain in the mother liquor. The subsequent deprotection step utilizes dilute hydrochloric acid in methanol at controlled low temperatures (0°C to 10°C) to cleave both the Boc group and the diphenylmethyl ester simultaneously. This tandem deprotection is kinetically controlled to prevent degradation of the sensitive cephem core, yielding the key 7-ACP intermediate with exceptional purity. Such rigorous control over reaction parameters and intermediate isolation is what defines a high-purity API intermediate process capable of meeting stringent pharmacopeial standards.
How to Synthesize Cefepime Hydrochloride Efficiently
The implementation of this synthesis route requires precise adherence to temperature controls and stoichiometric ratios to maximize yield and minimize impurity profiles. The process begins with the dissolution of the starting material in tetrahydrofuran, followed by the dropwise addition of the protecting agent under mild conditions. Detailed operational parameters, including specific molar ratios of triethylamine and Boc2O, are critical for ensuring complete conversion before proceeding to the quaternization stage. The subsequent steps involve careful solvent exchanges and temperature ramps during the acid cut and final acylation phases. For process chemists looking to replicate or license this technology, understanding the crystallization dynamics of the inner salt is paramount, as this step serves as the primary purification checkpoint before the final coupling reaction. The standardized synthetic steps outlined below provide a roadmap for achieving the high total yields reported in the patent examples.
- Protect the amino group of 7-amino-3-chloromethyl-3-cephem-4-carboxylic acid diphenylmethyl ester hydrochloride using di-tert-butyl dicarbonate in tetrahydrofuran.
- React the Boc-protected intermediate with N-methylpyrrolidine in dichloromethane to form the quaternary ammonium inner salt.
- Perform acid-mediated deprotection using methanol and dilute hydrochloric acid at controlled low temperatures to obtain 7-ACP.
- Conduct the final acylation reaction with AE-active ester in methanol followed by crystallization to yield high-purity Cefepime Hydrochloride.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, the adoption of this synthesis methodology offers profound advantages that extend beyond mere technical performance. The elimination of Freon and Carbon Tetrachloride from the supply chain removes a significant regulatory liability and reduces the complexity of waste management protocols. This transition to greener solvents like tetrahydrofuran and dichloromethane, which are widely available and easily recoverable through distillation, translates directly into substantial cost savings in terms of raw material procurement and hazardous waste disposal fees. Moreover, the simplified work-up procedures, which rely on crystallization rather than energy-intensive chromatography, reduce the overall processing time and utility consumption per kilogram of product. For supply chain heads, this means a more resilient production schedule that is less susceptible to disruptions caused by the scarcity of specialized reagents or tightening environmental regulations on solvent emissions.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of economically accessible raw materials and the recyclability of the solvent system. By avoiding the need for expensive, specialized fluorinated solvents and reducing the number of purification steps, the overall cost of goods sold (COGS) is significantly optimized. The high yield reported in the patent examples indicates a more efficient utilization of starting materials, reducing the financial loss associated with low-conversion batches. Additionally, the ability to perform tandem deprotection in a single step reduces reagent consumption and labor hours, further enhancing the cost-efficiency profile of the manufacturing operation.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as methanol, ether, and hydrochloric acid ensures that the supply chain remains robust and unaffected by the geopolitical or regulatory constraints often associated with controlled substances like Freon. The simplicity of the reaction conditions, which do not require extreme cryogenic temperatures or high-pressure equipment, allows for production in a wider range of facilities, thereby diversifying the potential manufacturing base. This flexibility is crucial for maintaining reducing lead time for high-purity API intermediates and ensuring continuous availability of critical antibiotic components even during market fluctuations.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, with reaction exotherms that are manageable and work-up procedures that are easily adapted to large-scale reactors. The significant reduction in three wastes (waste water, waste gas, and solid waste) aligns with global sustainability goals and corporate social responsibility mandates. By implementing a route that generates less hazardous byproduct and utilizes recyclable solvents, manufacturers can secure long-term operating licenses and avoid the reputational risks associated with environmental non-compliance, making it a sustainable choice for future production capacity expansion.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms legacy processes in terms of yield, purity, and operational safety. Understanding these details is vital for stakeholders evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: How does this synthesis method reduce isomer impurities compared to traditional routes?
A: By utilizing a Boc protection strategy on the 7-amino group prior to quaternization, the method prevents the nucleophilic attack of N-methylpyrrolidine on the beta-lactam ring, significantly suppressing the formation of delta-2 isomers common in unprotected pathways.
Q: What are the environmental advantages of this process over the Walker or Naito methods?
A: Unlike previous methods that required strictly prohibited polyhalogenated solvents like Freon or Carbon Tetrachloride, this novel route utilizes recyclable solvents such as tetrahydrofuran and dichloromethane, aligning with modern green chemistry standards and reducing hazardous waste disposal costs.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process features mild reaction conditions (25°C to 30°C), simple work-up procedures involving crystallization rather than complex chromatography, and high total yields up to 93%, making it highly robust for commercial scale-up of complex antibiotics.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cefepime Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthesis technologies to maintain competitiveness in the global pharmaceutical market. Our team of expert process chemists has thoroughly analyzed the pathway described in patent CN111171052B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this efficient route to life. We are committed to delivering stringent purity specifications through our rigorous QC labs, ensuring that every batch of Cefepime Hydrochloride or its key intermediates meets the highest international standards. Our state-of-the-art facilities are equipped to handle the specific solvent recovery and crystallization requirements of this green chemistry process, guaranteeing a consistent and high-quality supply for your downstream formulation needs.
We invite you to collaborate with us to leverage these technical advancements for your supply chain. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that demonstrate how our implementation of this novel synthesis method can drive value, reduce risk, and secure your position as a leader in the antibiotic market.