Advanced Synthetic Routes for High-Purity Adefovir Intermediates and Commercial Scalability
Advanced Synthetic Routes for High-Purity Adefovir Intermediates and Commercial Scalability
The pharmaceutical landscape for antiviral therapeutics continues to evolve, driven by the urgent need for efficient, safe, and scalable manufacturing processes for critical active pharmaceutical ingredients (APIs). Adefovir, chemically known as 9-(2-phosphonylmethoxyethyl)adenine, stands as a pivotal nucleoside analog used extensively in the treatment of hepatitis B virus infections. Its prodrug, Adefovir Dipivoxil, further underscores the commercial importance of this molecular scaffold. Recent intellectual property developments, specifically Patent CN100526321C, have unveiled a transformative synthetic methodology that addresses long-standing inefficiencies in the production of this vital compound. This patent details a robust pathway starting from 1,3-dioxolane, offering a distinct departure from traditional, hazard-prone routes. For R&D directors and procurement strategists alike, understanding the nuances of this innovation is crucial for securing a reliable pharmaceutical intermediates supplier capable of delivering high-purity materials with consistent quality.
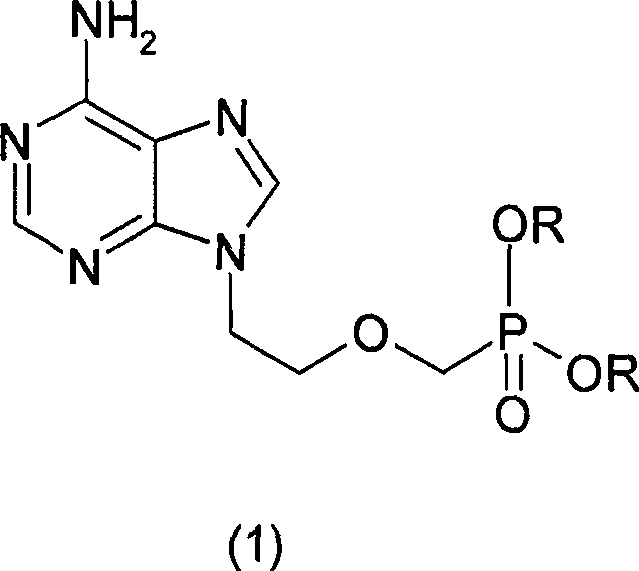
The structural complexity of Adefovir necessitates precise control over the phosphonate side chain installation. As illustrated in the chemical architecture, the molecule features a purine base linked to an acyclic phosphonate moiety. The challenge has historically lay in constructing this linkage without compromising the integrity of the adenine ring or introducing toxic impurities. The disclosed technology leverages a sequence involving ring-opening, rearrangement, and strategic alkylation to achieve this. By shifting the starting material paradigm away from volatile chloro-ethers towards stable cyclic acetals, the process inherently reduces the risk profile associated with the synthesis. This shift is not merely academic; it represents a tangible opportunity for cost reduction in pharmaceutical intermediates manufacturing by simplifying waste treatment protocols and enhancing worker safety standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Adefovir and its derivatives has relied heavily on routes originating from Virahol (isoprinosine) or direct alkylation strategies involving highly reactive and dangerous halogenated ethers. A predominant conventional method involves the reaction of Virahol with phosphorus trichloride to generate triisopropyl phosphite, followed by an Arbuzov reaction with 1-chloro-2-chloromethyl ethyl ether. While chemically feasible, this approach is fraught with significant operational drawbacks that hinder efficient commercial scale-up of complex pharmaceutical intermediates. The primary concern is the use of 1-chloro-2-chloromethyl ethyl ether, a substance identified as a potent mutagen with strong toxicity. Handling such genotoxic impurities requires specialized containment facilities, rigorous personal protective equipment, and extensive downstream purification to ensure the final API meets stringent regulatory limits. Furthermore, these traditional pathways often suffer from harsh reaction conditions that can lead to the degradation of sensitive intermediates, resulting in lower total yields and a complex impurity profile that complicates the purification process.
The Novel Approach
In stark contrast, the methodology outlined in Patent CN100526321C introduces a streamlined and safer alternative that fundamentally redesigns the construction of the side chain. This novel approach initiates with 1,3-dioxolane, a stable and commercially accessible cyclic acetal, reacting with acetyl chloride to form 2-chloromethane ethoxyacetic acid ethyl ester. This initial step sets the stage for a subsequent Arbuzov rearrangement with triisopropyl phosphite, effectively installing the phosphonate group under controlled thermal conditions. The brilliance of this route lies in its modularity and the mildness of its subsequent transformations. Following the rearrangement, the process employs a gentle hydrolysis step, potentially utilizing acidic ion exchange resins, to reveal the hydroxyl functionality required for the next coupling. This is followed by a chlorination step and finally, an N-alkylation with adenine. The entire sequence is designed to minimize by-product formation and operate under conditions that are far more forgiving than the corrosive and toxic environments of the prior art. This represents a significant leap forward for any organization seeking a reliable pharmaceutical intermediates supplier who prioritizes both quality and safety.
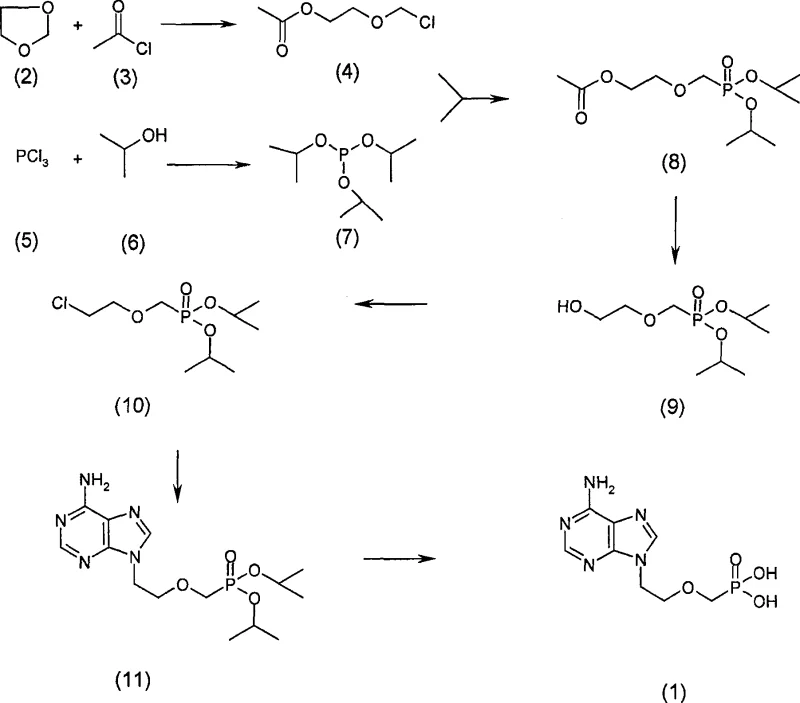
Mechanistic Insights into Arbuzov Rearrangement and N-Alkylation
To fully appreciate the technical merit of this synthesis, one must delve into the mechanistic underpinnings of the key transformation steps, particularly the Arbuzov rearrangement and the final deprotection. The reaction between the chloro-ester intermediate and triisopropyl phosphite is a classic example of the Michaelis-Arbuzov reaction, where a trivalent phosphorus species attacks an alkyl halide to form a quaternary phosphonium salt, which subsequently eliminates an alkyl halide to yield a pentavalent phosphonate. In this specific patent embodiment, the reaction is conducted at elevated temperatures (up to 125°C) to drive the equilibrium towards the desired phosphonate ester. The choice of isopropyl groups on the phosphorus serves a dual purpose: they provide sufficient steric bulk to stabilize the intermediate while remaining labile enough for removal in the final deprotection step. This balance is critical for maintaining high purity throughout the synthesis, as unstable intermediates can lead to difficult-to-remove phosphorous-containing impurities.
Furthermore, the final conversion of the protected intermediate (Compound 11) to the free acid Adefovir (Compound 1) showcases a sophisticated approach to deprotection. The patent describes the use of trimethylchlorosilane (TMSCl) in the presence of potassium iodide (KI) in acetonitrile. This reagent combination generates trimethylsilyl iodide (TMSI) in situ, a powerful silylating agent capable of cleaving phosphonate esters efficiently. The mechanism involves the silylation of the phosphonate oxygen, followed by nucleophilic attack by iodide to release the alkyl group as an alkyl iodide, leaving behind the free phosphonic acid. This method is superior to traditional acid hydrolysis which might require prolonged heating and strong mineral acids that could potentially degrade the adenine base. By optimizing these mechanistic steps, the process ensures that the final product exhibits the high-purity pharmaceutical intermediates characteristics required for clinical applications, with minimal risk of residual solvent or reagent contamination.
How to Synthesize Adefovir Efficiently
Implementing this synthetic route requires careful attention to reaction parameters and reagent quality to maximize yield and minimize impurities. The process begins with the preparation of the key chloro-ester building block, followed by the phosphonate installation and subsequent functional group manipulations. Each step, from the initial ring-opening of 1,3-dioxolane to the final crystallization of the API, is optimized for reproducibility and scalability. The use of inert gas protection throughout the reaction course is recommended to prevent oxidation of sensitive phosphorus species. For detailed operational procedures, including specific molar ratios, temperature profiles, and work-up techniques, refer to the standardized synthesis guide below which encapsulates the core teachings of the patent.
- React 1,3-dioxolane with acetyl chloride to form 2-chloromethane ethoxyacetic acid ethyl ester.
- Perform an Arbuzov rearrangement with triisopropyl phosphite to generate the phosphonate ester intermediate.
- Execute hydrolysis, chlorination, N-alkylation with adenine, and final deprotection to yield Adefovir.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers compelling economic and logistical benefits that extend beyond simple yield improvements. The elimination of mutagenic starting materials like 1-chloro-2-chloromethyl ethyl ether drastically simplifies the environmental, health, and safety (EHS) compliance burden. Facilities no longer need to invest in specialized containment systems for genotoxic compounds, nor do they face the high costs associated with the disposal of hazardous waste streams containing these toxins. This inherent safety profile translates directly into lower operational expenditures and reduced insurance liabilities. Moreover, the reliance on commodity chemicals such as 1,3-dioxolane and acetyl chloride ensures a stable and resilient supply chain, mitigating the risks of raw material shortages that often plague specialty chemical markets. This stability is essential for reducing lead time for high-purity pharmaceutical intermediates and ensuring continuous production schedules.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis allows for significant cost optimization through the reduction of processing steps and the avoidance of expensive purification protocols required to remove toxic impurities. By operating under milder conditions, the process also reduces energy consumption associated with extreme heating or cooling, contributing to a lower carbon footprint and reduced utility costs. The higher selectivity of the reactions means less raw material is wasted on by-products, improving the overall atom economy of the process. These factors combine to create a more economically viable manufacturing model that can withstand market fluctuations in raw material pricing.
- Enhanced Supply Chain Reliability: Sourcing raw materials that are widely available in the global chemical market reduces dependency on single-source suppliers of niche reagents. The robustness of the reaction conditions means that the process is less susceptible to minor variations in feedstock quality, ensuring consistent output even when supply chains are stressed. This reliability is critical for maintaining the continuity of API production, preventing costly downtime, and meeting the rigorous delivery schedules demanded by downstream pharmaceutical partners. A stable supply of intermediates ensures that the final drug product can reach patients without interruption.
- Scalability and Environmental Compliance: The chemistry described is inherently scalable, having been designed with industrial application in mind. The use of standard unit operations such as distillation, filtration, and crystallization facilitates easy technology transfer from pilot plant to commercial scale. Additionally, the reduced generation of hazardous waste aligns with increasingly stringent global environmental regulations, future-proofing the manufacturing site against tighter emission standards. This proactive approach to environmental compliance not only avoids potential fines but also enhances the corporate reputation of the manufacturer as a responsible partner in the healthcare value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced Adefovir synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the practical implications for potential manufacturing partners. Understanding these details is key to evaluating the feasibility of adopting this route for your specific production needs.
Q: What are the safety advantages of the new Adefovir synthesis route?
A: The novel route avoids the use of 1-chloro-2-chloromethyl ethyl ether, a known mutagen used in conventional methods, thereby significantly improving operator safety and reducing hazardous waste handling requirements.
Q: How does the new method improve yield compared to traditional processes?
A: By utilizing milder reaction conditions and specific catalysts like acidic ion exchange resins for hydrolysis, the process minimizes by-product formation and decomposition, leading to superior overall recovery of the target nucleoside analog.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process utilizes readily available raw materials like 1,3-dioxolane and operates under manageable temperature ranges (-10°C to 200°C), making it highly adaptable for commercial scale-up without requiring exotic high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Adefovir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting cutting-edge synthetic methodologies to meet the evolving demands of the global pharmaceutical industry. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovations like the Adefovir route described in Patent CN100526321C can be seamlessly translated into reality. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that employ state-of-the-art analytical techniques to verify every batch. Our dedication to quality assurance ensures that every intermediate we supply meets the exacting standards required for GMP manufacturing of final drug substances.
We invite you to collaborate with us to leverage these technological advancements for your supply chain. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise can drive efficiency and reliability in your Adefovir production strategy. Together, we can build a more sustainable and cost-effective future for antiviral therapeutics.