Advanced Synthesis of Candesartan Cilexetil Intermediates via Optimized Esterification and Deprotection
The pharmaceutical industry continuously seeks robust and scalable methodologies for the production of antihypertensive agents, particularly Angiotensin II receptor antagonists like Candesartan Cilexetil. A pivotal advancement in this domain is detailed in patent CN101817794A, which discloses a highly efficient preparation technology for a key intermediate in the Candesartan Cilexetil synthesis pathway. This innovation addresses critical bottlenecks in traditional manufacturing by introducing a mild, safe, and operationally simple protocol that is exceptionally well-suited for large-scale industrial application. The core of this technology lies in a refined two-step sequence involving a specialized esterification followed by a selective deprotection, ensuring high yields while minimizing the formation of hazardous byproducts. By optimizing reaction conditions and solvent systems, this process offers a compelling value proposition for manufacturers aiming to enhance the reliability of their supply chains for cardiovascular medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Candesartan Cilexetil intermediates has been plagued by several inefficiencies that hinder cost-effective mass production. Traditional routes often rely on high-boiling polar aprotic solvents such as dimethylformamide (DMF), which pose significant challenges during the workup phase due to their difficulty in removal and high energy requirements for distillation. Furthermore, conventional deprotection strategies frequently involve harsh conditions or multiple discrete steps to remove different protecting groups, increasing the risk of damaging sensitive functional groups like the carbonate ester side chain. These multi-step processes not only extend the overall lead time but also accumulate impurities, necessitating rigorous and costly purification procedures such as column chromatography at intermediate stages. The cumulative effect of these limitations is a process with lower atom economy, higher waste generation, and reduced throughput, making it less attractive for modern, green chemistry-focused manufacturing environments.
The Novel Approach
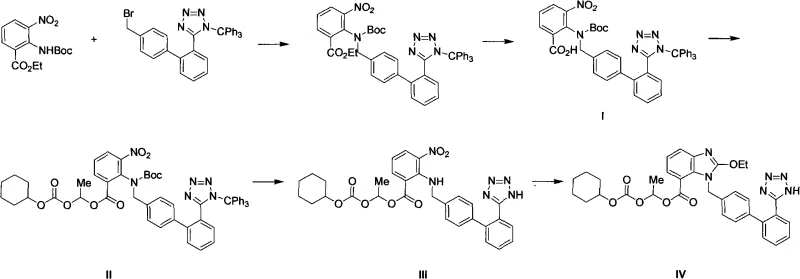
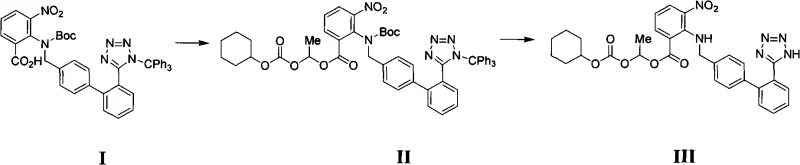
In stark contrast, the methodology outlined in the patent introduces a streamlined approach that fundamentally alters the process landscape for producing these critical intermediates. The novel route utilizes lower-boiling solvents like acetonitrile or tetrahydrofuran (THF) for the initial esterification, which dramatically simplifies solvent recovery and reduces the thermal load on the reaction mixture. As illustrated in the reaction scheme below, the process converts Compound I directly to Compound II through an efficient alkylation, followed by a sophisticated one-pot deprotection to yield Compound III. This approach eliminates the need for intermediate purification, allowing the crude product from the first step to proceed directly to the second. The ability to remove both N-Boc and N-Trityl protecting groups simultaneously under mild acidic conditions without compromising the integrity of the ester side chain represents a significant leap in chemoselectivity and process intensification.
Mechanistic Insights into Alkaline Esterification and Acidic Deprotection
The success of this synthesis hinges on the precise control of reaction parameters during the alkaline esterification phase. In this step, Compound I reacts with 1-chloroethyl cyclohexyl carbonate in the presence of a base such as potassium carbonate (K2CO3) or sodium carbonate (Na2CO3). The choice of base is critical; it must be strong enough to deprotonate the carboxylic acid moiety to facilitate nucleophilic attack on the chloro-carbonate, yet mild enough to prevent hydrolysis of the sensitive ester linkages. The reaction proceeds optimally at temperatures ranging from 20°C to 80°C, with reflux in acetonitrile proving particularly effective. This temperature window ensures sufficient kinetic energy for the substitution reaction while avoiding thermal degradation. The mechanism involves the formation of a carboxylate anion which attacks the electrophilic carbon of the chloro-carbonate, displacing the chloride ion and forming the desired mixed carbonate ester linkage found in Compound II.
Following the esterification, the deprotection mechanism is equally sophisticated, designed to achieve orthogonality in protecting group removal. The process employs an acidic alcoholic solution, potentially generated in situ using acetyl chloride and ethanol or simply using HCl in ethanol. The acid catalyzes the cleavage of the tert-butyloxycarbonyl (Boc) group via the formation of a carbocation intermediate, which subsequently eliminates isobutylene. Simultaneously, the triphenylmethyl (Trityl) group on the tetrazole ring is protonated and cleaved as a stable trityl cation. The brilliance of this system lies in its selectivity; the conditions are tuned such that the 1-[[(cyclohexyloxy)carbonyl]oxy]ethyl ester side chain remains stable. This stability is crucial because hydrolysis of this side chain would lead to inactive impurities. By maintaining the temperature between -10°C and 50°C, the reaction kinetics favor the removal of the nitrogen protecting groups while preserving the oxygen-based ester functionality, resulting in a high-purity final intermediate.
How to Synthesize Candesartan Cilexetil Intermediate Efficiently
Implementing this synthesis requires careful attention to reagent quality and temperature control to maximize the benefits of the patented process. The procedure begins with the suspension of the starting acid and base in the chosen organic solvent, followed by the controlled addition of the chloro-carbonate reagent. Monitoring the reaction via TLC is essential to determine the precise endpoint before proceeding to the workup, which involves simple filtration and solvent evaporation. The resulting crude oil is then subjected to the acidic deprotection conditions, where temperature ramping from ice-bath conditions to room temperature allows for a controlled release of protecting groups. For a comprehensive, step-by-step guide including specific molar ratios, stirring rates, and safety protocols, please refer to the standardized synthesis instructions provided below.
- Perform alkaline esterification of Compound I with 1-chloroethyl cyclohexyl carbonate in acetonitrile or THF at 20-80°C using K2CO3 or Na2CO3.
- Isolate Compound II crude product by filtration and solvent concentration without further purification.
- Conduct acidic deprotection of Compound II in alcoholic solution (e.g., ethanol with acetyl chloride or HCl) at -10 to 50°C to remove Boc and Trityl groups, yielding Compound III.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this technology translates into tangible strategic advantages that go beyond mere technical feasibility. The shift away from high-boiling solvents like DMF to more volatile and easily recoverable solvents like acetonitrile significantly reduces the energy burden associated with solvent recycling and waste treatment. This modification not only lowers utility costs but also accelerates the batch cycle time, allowing for greater throughput within existing infrastructure. Furthermore, the elimination of intermediate purification steps means a drastic reduction in the consumption of silica gel, eluents, and labor hours, directly impacting the cost of goods sold (COGS). The robustness of the reaction conditions also implies a lower risk of batch failure, ensuring a more consistent and reliable supply of critical intermediates for downstream API manufacturing.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the simplification of the workflow and the optimization of material usage. By enabling the direct use of crude Compound II in the subsequent step, the process removes an entire unit operation dedicated to purification, which typically accounts for a significant portion of manufacturing expenses. Additionally, the replacement of expensive or difficult-to-handle solvents with commodity chemicals like acetonitrile and ethanol reduces raw material procurement costs. The high selectivity of the deprotection step minimizes the formation of hard-to-remove impurities, thereby reducing the yield loss often associated with aggressive purification attempts. These factors combine to create a leaner, more cost-efficient manufacturing profile that enhances competitiveness in the generic pharmaceutical market.
- Enhanced Supply Chain Reliability: From a supply chain perspective, the reliance on readily available and stable reagents such as potassium carbonate, acetyl chloride, and common alcohols mitigates the risk of raw material shortages. Unlike specialized catalysts or exotic reagents that may have long lead times or single-source dependencies, the inputs for this process are commoditized and widely accessible globally. The mild reaction conditions also reduce the wear and tear on reactor vessels and ancillary equipment, leading to lower maintenance downtime and higher asset utilization rates. This operational stability ensures that production schedules can be met consistently, providing downstream partners with the confidence of uninterrupted supply for their finished dosage form production lines.
- Scalability and Environmental Compliance: The design of this synthesis inherently supports scalability, a critical factor for meeting the growing global demand for antihypertensive medications. The exothermic nature of the reactions is manageable within standard jacketed reactors, and the absence of cryogenic requirements or extreme pressures simplifies the engineering controls needed for scale-up. Environmentally, the process aligns with green chemistry principles by reducing solvent waste and avoiding the generation of heavy metal contaminants often associated with alternative catalytic methods. The ease of solvent recovery further diminishes the environmental footprint, facilitating compliance with increasingly stringent regulatory standards regarding volatile organic compound (VOC) emissions and hazardous waste disposal.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of integrating this technology into their existing portfolios, we have compiled answers to common inquiries regarding the process specifics and quality outcomes. These responses are derived directly from the experimental data and technical disclosures within the patent documentation, ensuring accuracy and relevance for process development scientists. Understanding these nuances is essential for conducting a thorough risk assessment and for planning the necessary analytical method validations required for regulatory filings. We encourage stakeholders to review these points as a foundation for deeper technical discussions with our engineering team.
Q: What are the key advantages of using acetonitrile over DMF in this synthesis?
A: Using acetonitrile replaces high-boiling solvents like DMF, significantly simplifying the post-reaction workup. It allows for easier solvent removal via distillation, reduces energy consumption, and minimizes thermal stress on the sensitive intermediate, thereby improving overall process safety and efficiency.
Q: How does the new deprotection method improve selectivity?
A: The novel acidic deprotection conditions (e.g., Acetyl Chloride/Ethanol or HCl/Ethanol) allow for the simultaneous removal of both N-Boc and N-Trityl protecting groups in a single step. Crucially, this method maintains high chemoselectivity, leaving the sensitive 1-[[(cyclohexyloxy)carbonyl]oxy]ethyl ester side chain intact, which reduces side reactions and impurities.
Q: Is intermediate purification required between steps?
A: No, one of the significant operational advantages of this technology is that the crude Compound II obtained after the esterification step can be directly used in the subsequent deprotection reaction without column chromatography or recrystallization. This telescoping of steps drastically reduces processing time and solvent usage.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Candesartan Cilexetil Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. Our facility is equipped to handle complex organic syntheses, including the multi-step pathways required for advanced cardiovascular intermediates like those described in CN101817794A. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify every batch against the highest industry standards.
We invite you to collaborate with us to leverage this optimized synthesis technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and logistical constraints. By partnering with us, you gain access not only to high-quality intermediates but also to valuable insights on route feasibility assessments and specific COA data that can accelerate your regulatory approval timelines. Contact us today to discuss how we can support your supply chain goals with reliable, cost-effective, and compliant chemical solutions.