Industrial Synthesis of Candesartan Intermediate: A Breakthrough in Scalable API Manufacturing
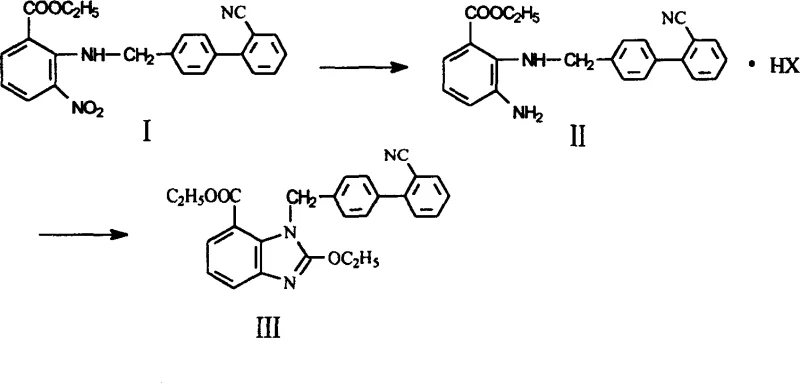
The pharmaceutical industry continuously seeks robust synthetic pathways that balance high purity with economic viability, particularly for critical antihypertensive agents like Candesartan Cilexetil. Patent CN1272325C introduces a transformative preparation method for the key intermediate 1-((2'-cyanobiphenyl-4-yl)methyl)-2-ethoxylbenzimidazole-7-ethyl formate, addressing long-standing inefficiencies in prior art. This innovation shifts the paradigm from laboratory-scale purification techniques to a streamlined industrial process, leveraging tin dichloride reduction followed by a novel salt formation strategy. By fundamentally altering the isolation mechanism of the amino intermediate, this technology offers a reliable pharmaceutical intermediate supplier pathway that drastically reduces operational complexity. The significance of this patent lies not merely in chemical novelty but in its direct applicability to commercial scale-up of complex intermediates, ensuring consistent quality and supply continuity for global API manufacturers seeking competitive advantages.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for this benzimidazole derivative have historically relied heavily on labor-intensive and solvent-heavy purification protocols that hinder industrial scalability. Prior art methods typically involve reducing the nitro precursor followed by rigorous silica-gel column chromatography to isolate the oily amino intermediate, a process fraught with variability and material loss. Furthermore, subsequent recrystallization using ethyl acetate-hexane mixtures adds another layer of complexity, requiring precise solvent recovery systems and extensive processing time. These conventional steps are not only costly due to the high consumption of chromatographic media and organic solvents but also pose significant challenges in waste management and environmental compliance. The cumulative yield of such multi-step purification often drops precipitously, with historical data indicating overall yields as low as 21.3%, rendering the process economically unsustainable for large-volume production. Consequently, manufacturers face inflated production costs and extended lead times, creating bottlenecks in the supply chain for high-purity pharmaceutical intermediates.
The Novel Approach
In stark contrast, the patented methodology introduces a sophisticated salt formation technique that bypasses the need for chromatographic purification entirely, marking a significant leap in process chemistry efficiency. Instead of isolating the amino intermediate as an oil through column chromatography, the process utilizes an acidic organic solvent system to convert the reduced product directly into a solid salt. This solid salt can be easily filtered and washed, effectively purifying the compound through crystallization rather than adsorption, which significantly enhances recovery rates. The subsequent cyclization step proceeds directly from this solid salt using tetraethyl orthocarbonate under glacial acetic acid conditions, streamlining the workflow into a cohesive, continuous operation. This approach not only simplifies the operational protocol but also aligns perfectly with the requirements for cost reduction in API manufacturing by minimizing solvent usage and eliminating expensive stationary phases. The result is a robust, scalable process that transforms a previously cumbersome laboratory procedure into a viable industrial manufacturing route.
Mechanistic Insights into Tin Dichloride Reduction and Salt-Mediated Cyclization
The core of this technological advancement lies in the precise control of the reduction and salification steps, which dictate the purity and physical form of the intermediate. The reduction of the nitro group using tin dichloride is conducted in an alcoholic solvent, where the resulting amine is immediately subjected to an acidic environment containing 10 to 40% W/W acid. This specific acidity range is critical for protonating the amine functionality, thereby increasing its polarity and reducing its solubility in the organic phase, which drives the precipitation of the solid salt. By controlling the acid concentration and solvent composition, such as using ethanol or isopropanol mixed with hydrochloric or hydrobromic acid, the process ensures that impurities remain in the supernatant while the desired product crystallizes out. This mechanism effectively replaces the separation power of silica gel with thermodynamic solubility differences, offering a more predictable and controllable purification outcome.
Following the isolation of the solid salt, the cyclization mechanism involves the reaction of the amino ester with tetraethyl orthocarbonate in the presence of glacial acetic acid. The acidic conditions facilitate the nucleophilic attack of the amine on the orthoester, leading to the formation of the benzimidazole ring with the elimination of ethanol. The use of the pre-formed salt ensures that the amine is in a reactive state that favors cyclization over side reactions, thereby enhancing the selectivity of the transformation. This step is crucial for maintaining the structural integrity of the biphenyl moiety and the ester functionality, which are essential for the biological activity of the final drug substance. The elimination of transition metal catalysts or harsh reagents in this step further contributes to a cleaner impurity profile, reducing the burden on downstream purification processes.
How to Synthesize 1-((2'-cyanobiphenyl-4-yl)methyl)-2-ethoxylbenzimidazole-7-ethyl formate Efficiently
Implementing this synthesis requires strict adherence to the patented parameters regarding acid concentration and solvent selection to maximize yield and purity. The process begins with the reduction of the nitro precursor, followed by the critical salification step where the choice of acid and solvent ratio determines the efficiency of precipitation. Operators must ensure that the reaction mixture is concentrated appropriately before the addition of the acidic solution to induce optimal crystal formation. Detailed standardized synthetic steps see the guide below, which outlines the specific temperature profiles and stoichiometric ratios required for reproducible results. Adhering to these guidelines ensures that the transition from laboratory bench to pilot plant is seamless, maintaining the high yield improvements documented in the patent literature.
- Reduce 3-nitro-2-(((2'-cyanobiphenyl-4-yl)methyl)amino)ethyl benzoate using tin dichloride in an alcoholic solvent at elevated temperatures.
- Add an acidic solution to the reaction mixture to form a solid salt of the amino intermediate, precipitating it directly from the organic phase.
- React the isolated solid salt with tetraethyl orthocarbonate under glacial acetic acid conditions to cyclize and form the final benzimidazole derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented process translates into tangible strategic benefits that extend beyond simple chemical yield improvements. By eliminating the reliance on silica gel chromatography, the manufacturing process becomes significantly less dependent on specialized consumables that are subject to market volatility and supply constraints. This shift allows for a more predictable production schedule, as the bottlenecks associated with column packing, running, and regeneration are completely removed from the workflow. Furthermore, the reduction in solvent volume and the simplification of unit operations lead to substantial cost savings in terms of energy consumption and waste disposal fees. These efficiencies enable a reliable pharmaceutical intermediate supplier to offer more competitive pricing structures while maintaining healthy margins, ultimately strengthening the resilience of the global supply chain for antihypertensive medications.
- Cost Reduction in Manufacturing: The removal of silica-gel column chromatography and ethyl acetate-hexane recrystallization eliminates two of the most expensive and labor-intensive steps in the traditional synthesis. Without the need for large quantities of chromatographic silica and the associated solvent recovery infrastructure, the variable cost per kilogram of the intermediate is drastically lowered. Additionally, the higher overall yield means that less starting material is required to produce the same amount of final product, further compounding the raw material savings. This economic efficiency allows manufacturers to absorb fluctuations in raw material prices more effectively, ensuring stable pricing for downstream API producers.
- Enhanced Supply Chain Reliability: Simplifying the synthesis route reduces the number of potential failure points in the manufacturing process, leading to higher batch success rates and consistent delivery timelines. The use of common industrial solvents and acids, rather than specialized purification media, ensures that the supply chain is not vulnerable to shortages of niche chemicals. This robustness is critical for maintaining continuous production schedules, especially when scaling up to meet the demands of large-scale commercial contracts. By adopting this method, companies can reduce lead time for high-purity pharmaceutical intermediates, providing a competitive edge in fast-moving markets.
- Scalability and Environmental Compliance: The process is inherently designed for large-scale industrial production, avoiding the scalability issues inherent in chromatographic separations which are difficult to translate from gram to ton scale. The reduction in solvent usage and the elimination of silica waste contribute to a smaller environmental footprint, aligning with increasingly stringent global regulations on chemical manufacturing emissions. This environmental compliance reduces the risk of regulatory shutdowns and facilitates easier permitting for new production facilities. Consequently, the technology supports sustainable growth and long-term viability for manufacturers committed to green chemistry principles.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent documentation, providing clarity for R&D and procurement teams evaluating this technology. Understanding these details is essential for assessing the feasibility of integrating this process into existing manufacturing lines or for qualifying new suppliers. The insights provided here aim to bridge the gap between theoretical patent claims and practical industrial application.
Q: How does this patent improve the yield compared to traditional methods?
A: The patented method increases the overall yield from approximately 21.3% in conventional routes to 65.8% by eliminating purification losses associated with silica gel column chromatography.
Q: Does this process require expensive chromatography equipment?
A: No, the process explicitly abolishes the need for silica-gel column chromatography and ethyl acetate-hexane recrystallization, relying instead on salt formation for purification.
Q: Is this synthesis suitable for large-scale commercial production?
A: Yes, the method is specifically designed for industrial suitability by simplifying operations, reducing solvent usage, and avoiding labor-intensive purification steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Candesartan Cilexetil Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in maintaining a competitive edge in the global pharmaceutical market. Our team of expert chemists has extensively evaluated the patented process described in CN1272325C and possesses the technical capability to implement this advanced methodology at scale. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the benefits of this high-yield route are fully realized in a GMP-compliant environment. Our rigorous QC labs and stringent purity specifications guarantee that every batch of intermediate meets the exacting standards required for API synthesis, providing our partners with peace of mind and supply security.
We invite forward-thinking pharmaceutical companies to collaborate with us to optimize their supply chains and reduce manufacturing costs through the adoption of this superior technology. By leveraging our expertise, you can access a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, allowing you to make informed decisions that drive value and efficiency in your drug development pipeline. Together, we can redefine the standards of excellence in intermediate manufacturing.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →