Advanced Catalytic Synthesis of Trifluoromethyl Dihydrofuran Amines for Commercial Scale-Up
Introduction to Next-Generation Fluorinated Heterocycles
The integration of fluorine atoms into heterocyclic scaffolds represents a cornerstone strategy in modern medicinal chemistry, particularly for enhancing metabolic stability and bioavailability in drug candidates. Patent CN110922369B introduces a groundbreaking methodology for the synthesis of trifluoromethyl-substituted dihydrofuran amine compounds, addressing critical gaps in current organic synthesis capabilities. This technology enables the efficient construction of 2,3-dihydrofuran rings bearing a quaternary carbon stereocenter, a structural motif that is notoriously difficult to access via traditional routes. The significance of this innovation lies in its ability to transform simple, acyclic precursors into complex, highly functionalized heterocycles under remarkably mild conditions. By leveraging a copper-catalyzed cyclization strategy, this approach bypasses the need for harsh reagents, thereby expanding the scope of compatible substrates to include sensitive pharmacophores essential for advanced pharmaceutical development.
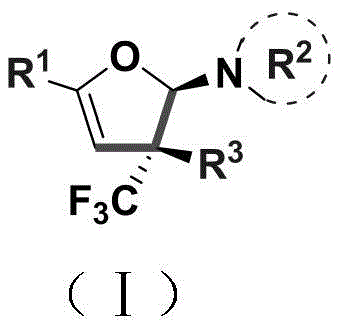
Furthermore, the versatility of this synthetic platform extends beyond mere ring formation; it provides a robust entry point for generating diverse libraries of fluorinated intermediates. The resulting dihydrofuran amines are not merely end-products but serve as pivotal synthons for downstream transformations, including the preparation of trifluoromethyl-containing 1,4-dicarbonyl compounds. For R&D directors seeking reliable sources of high-purity pharmaceutical intermediates, this patent outlines a pathway that combines structural complexity with operational simplicity. The ability to install a trifluoromethyl group directly during the cyclization event eliminates multiple synthetic steps, streamlining the overall production timeline and reducing the cumulative waste associated with multi-step sequences. This positions the technology as a vital asset for companies aiming to accelerate their pipeline of fluorinated agrochemical and pharmaceutical agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of dihydrofuran rings, especially those substituted with trifluoromethyl groups, has been plagued by significant synthetic hurdles that impede large-scale manufacturing. Conventional strategies often necessitate the use of stoichiometric quantities of transition metals, which not only drives up raw material costs but also creates substantial challenges in downstream purification due to heavy metal contamination. Moreover, many established protocols require extreme reaction conditions, such as high temperatures or strongly acidic/basic environments, which are incompatible with the delicate functional groups frequently found in biologically active molecules. These harsh parameters often lead to poor yields, decomposition of sensitive substrates, and limited scope regarding electronic diversity on the aromatic rings. Additionally, the one-pot introduction of a CF3 group alongside ring closure from acyclic precursors has remained a rare and difficult transformation, typically requiring specialized reagents that are neither cost-effective nor readily available for commercial scale-up.
The Novel Approach
In stark contrast to these legacy issues, the methodology disclosed in CN110922369B utilizes a catalytic system that operates under温和 (mild) conditions, fundamentally shifting the paradigm for accessing these valuable scaffolds. The process employs a mere 5 mol% of copper(II) chloride as a catalyst, drastically reducing the metal load compared to stoichiometric alternatives and simplifying the removal of metal residues. By reacting an enaminone (Compound A) with a trifluoromethylhydrazone (Compound B) in the presence of potassium tert-butoxide, the reaction proceeds smoothly at temperatures between 80°C and 100°C. This thermal window is easily achievable in standard industrial reactors without requiring specialized high-pressure or cryogenic equipment. The result is a highly efficient cyclization that tolerates a wide array of substituents, including electron-withdrawing nitro and cyano groups as well as electron-donating methoxy moieties, ensuring broad applicability across different chemical spaces.
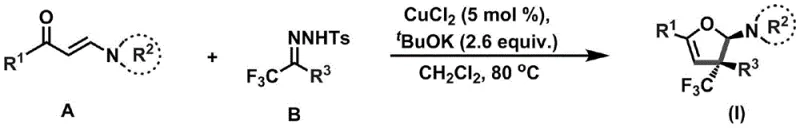
The operational simplicity of this novel approach translates directly into tangible benefits for procurement and supply chain management. The use of dichloromethane as a solvent, combined with a straightforward workup procedure involving filtration and column chromatography, minimizes the complexity of the manufacturing process. Unlike older methods that might require multiple protection-deprotection sequences or hazardous reagents, this one-pot transformation consolidates bond formation and functional group installation into a single step. This consolidation not only reduces the total number of unit operations but also enhances the overall atom economy of the synthesis. For manufacturers targeting cost reduction in pharmaceutical intermediate manufacturing, this efficiency gain is critical, as it lowers both the direct material costs and the indirect overheads associated with prolonged reaction times and complex purification trains.
Mechanistic Insights into CuCl2-Catalyzed Cyclization
From a mechanistic perspective, the success of this transformation relies on the synergistic interaction between the copper catalyst and the base to activate the hydrazone precursor. The copper(II) chloride likely acts as a Lewis acid, coordinating with the nitrogen atoms of the trifluoromethylhydrazone to facilitate the generation of a reactive diazo or carbene-like species in situ. Simultaneously, the potassium tert-butoxide serves to deprotonate the enaminone, increasing its nucleophilicity and enabling a concerted attack on the activated trifluoromethyl species. This dual activation strategy ensures that the cyclization proceeds with high regioselectivity and stereoselectivity, crucial for establishing the quaternary carbon center at the 3-position of the dihydrofuran ring. The preservation of stereochemistry is paramount for pharmaceutical applications, where the biological activity of a drug candidate is often strictly dependent on its three-dimensional configuration. The robustness of this catalytic cycle allows for the consistent production of a single stereoisomer, reducing the need for costly chiral separations later in the synthesis.
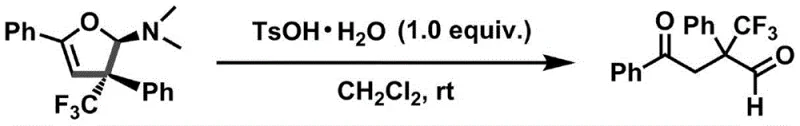
Furthermore, the mechanism inherently supports excellent functional group tolerance, a key requirement for late-stage functionalization in drug discovery. The mild basic conditions and the specific nature of the copper catalyst prevent the degradation of sensitive groups such as halides (fluoro, chloro, bromo) and esters, which might otherwise undergo hydrolysis or elimination under harsher regimes. This tolerance extends to the subsequent derivatization of the dihydrofuran amine product. As demonstrated in the patent, the amine moiety can be hydrolyzed under acidic conditions using p-toluenesulfonic acid to yield trifluoromethyl-containing 1,4-dicarbonyl compounds. This secondary transformation opens up additional avenues for constructing complex heterocycles, such as pyrroles or furans, further amplifying the utility of the initial cyclization product. The ability to seamlessly transition from the cyclic amine to open-chain dicarbonyls provides chemists with a flexible toolkit for exploring diverse chemical space.
How to Synthesize Trifluoromethyl Dihydrofuran Amine Efficiently
The practical implementation of this synthesis is designed for reproducibility and ease of handling, making it suitable for both laboratory-scale optimization and pilot-plant production. The protocol begins with the precise weighing of the enaminone and trifluoromethylhydrazone substrates, followed by the addition of the catalytic copper salt and the stoichiometric base. Maintaining an inert atmosphere, typically using argon, is recommended to prevent oxidation of sensitive intermediates, although the reaction demonstrates reasonable stability. The mixture is then heated to the optimal range of 80-100°C, where it is stirred for a period of 48 to 72 hours to ensure full conversion. Monitoring the reaction progress via TLC or HPLC is advised to determine the exact endpoint, preventing over-reaction or decomposition. Once the reaction is complete, the workup is remarkably straightforward, involving a simple filtration to remove inorganic salts followed by solvent evaporation.
- Mix enaminone (Compound A), trifluoromethylhydrazone (Compound B), CuCl2 (5 mol%), and tBuOK (2.6 equiv.) under inert gas protection.
- Add dichloromethane solvent and heat the reaction mixture to 80-100°C, stirring for 48-72 hours to ensure complete conversion.
- Filter insoluble solids, concentrate the filtrate under reduced pressure, and purify the crude residue via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented technology offers a compelling value proposition centered around risk mitigation and cost efficiency. The shift from stoichiometric to catalytic metal usage represents a significant reduction in raw material expenditure, particularly given the volatility of transition metal prices. Moreover, the simplified purification process reduces the consumption of silica gel and solvents, which are major cost drivers in fine chemical manufacturing. The mild reaction conditions also enhance operational safety, lowering the energy requirements for heating and cooling and reducing the risk of thermal runaways that can disrupt production schedules. These factors collectively contribute to a more resilient supply chain capable of meeting the rigorous demands of the global pharmaceutical market without compromising on quality or delivery timelines.
- Cost Reduction in Manufacturing: The utilization of a catalytic amount of copper chloride (5 mol%) instead of stoichiometric transition metals drastically lowers the cost of goods sold (COGS) by minimizing expensive metal waste. Additionally, the elimination of complex multi-step sequences in favor of a one-pot cyclization reduces labor costs and facility occupancy time. The streamlined workup procedure, which avoids intricate extraction or distillation steps, further decreases utility consumption and waste disposal fees. This holistic reduction in process complexity translates into substantial cost savings that can be passed down to the customer or reinvested into R&D initiatives.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions ensures high batch-to-batch consistency, a critical factor for maintaining reliable inventory levels. The tolerance for a wide variety of substrates means that supply chain disruptions for specific starting materials can be mitigated by switching to alternative analogs without re-optimizing the entire process. Furthermore, the use of common solvents like dichloromethane and readily available reagents like potassium tert-butoxide ensures that raw material sourcing remains stable and unaffected by niche market fluctuations. This reliability is essential for long-term contracts with multinational corporations that demand uninterrupted supply.
- Scalability and Environmental Compliance: The process is inherently scalable, as the exothermic profile is manageable and the reaction does not require specialized high-pressure equipment. This facilitates a smooth transition from gram-scale laboratory synthesis to ton-scale commercial production. From an environmental perspective, the reduced metal load simplifies wastewater treatment and lowers the burden of heavy metal compliance, aligning with increasingly stringent global environmental regulations. The high atom economy of the cyclization also minimizes the generation of organic waste, supporting sustainability goals and reducing the carbon footprint of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the feasibility and advantages of the method. Understanding these details is crucial for technical teams evaluating the integration of this route into their existing manufacturing portfolios. The responses highlight the balance between scientific rigor and practical applicability that defines this innovation.
Q: What are the limitations of conventional dihydrofuran synthesis methods?
A: Traditional methods often rely on stoichiometric amounts of transition metals, require harsh reaction conditions that limit functional group tolerance, and struggle to introduce trifluoromethyl groups efficiently from simple acyclic substrates.
Q: How does the novel Cu-catalyzed method improve stereoselectivity?
A: The patented process effectively constructs a quaternary carbon stereocenter with high stereoselectivity, utilizing mild conditions that preserve sensitive functional groups while ensuring the robust formation of the 2,3-dihydrofuran core.
Q: Can this intermediate be further derivatized for drug discovery?
A: Yes, the resulting dihydrofuran amines serve as versatile precursors that can be quantitatively converted into trifluoromethyl-containing 1,4-dicarbonyl compounds, which are valuable building blocks for constructing complex heterocyclic drug molecules.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Dihydrofuran Amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic route described in CN110922369B for advancing fluorinated drug candidates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our state-of-the-art facilities are equipped to handle the specific requirements of copper-catalyzed reactions, including rigorous QC labs that enforce stringent purity specifications to meet the highest industry standards. We understand that the integrity of your supply chain depends on our ability to deliver high-purity intermediates consistently, and we are committed to being a dependable extension of your own production capabilities.
We invite you to engage with our technical procurement team to discuss how this technology can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of adopting this catalytic route for your specific targets. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing us to demonstrate our commitment to quality and transparency. Together, we can accelerate the development of next-generation therapeutics by leveraging cutting-edge synthetic methodologies that prioritize efficiency, safety, and scalability.