Advanced Manufacturing of Cefotiam Hexetil Hydrochloride for Global Pharmaceutical Supply Chains
The pharmaceutical industry continuously seeks robust synthetic routes for second-generation cephalosporins, particularly for oral formulations that offer superior bioavailability. Patent CN101948476B introduces a groundbreaking methodology for the preparation and purification of cefotiam hexetil hydrochloride, a critical prodrug that hydrolyzes rapidly into the active cefotiam upon absorption. This innovation addresses long-standing challenges in the synthesis of this antibiotic intermediate, specifically targeting the issues of low yield, high isomer content, and product discoloration that have plagued traditional manufacturing protocols. By shifting from harsh purification techniques to a refined combination of extraction and crystallization, this technology enables the production of high-purity intermediates essential for modern generic and branded drug development.
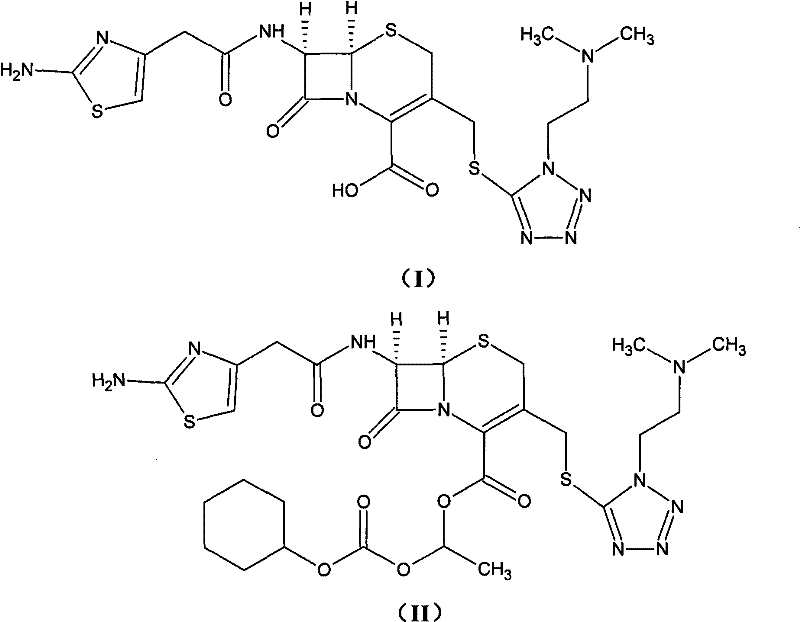
For procurement specialists and supply chain directors, the implications of this patent extend beyond mere chemical curiosity; it represents a tangible opportunity for cost reduction in antibiotic intermediate manufacturing. The ability to consistently achieve purity levels exceeding 98.5% with yields greater than 95% drastically reduces the cost of goods sold (COGS) by minimizing raw material waste and downstream processing time. As a reliable cefotiam hexetil hydrochloride supplier, understanding these mechanistic advantages allows us to offer partners a more stable and economically viable supply chain, mitigating the risks associated with volatile production yields and complex purification bottlenecks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of cefotiam hexetil has been fraught with inefficiencies that hinder large-scale commercial viability. Prior art methods, such as those described in early literature involving the reaction of cefotiam potassium with 1-iodoethyl cyclohexyl carbonate in DMF, often suffered from critically low yields, sometimes dropping as low as 20%. Furthermore, these conventional routes struggled to control the formation of the Δ2 isomer, frequently resulting in impurity levels that exceeded the stringent limits set by pharmacopoeias like the Japanese Pharmacopoeia. The reliance on column chromatography for purification not only increased operational complexity but also introduced significant product loss, while the tendency of the product to develop pink or faint yellow hues necessitated additional decolorization steps using activated carbon, further complicating the isolation process.
The Novel Approach
The methodology disclosed in CN101948476B offers a transformative solution by re-engineering the reaction pathway to favor high conversion and selectivity. Instead of relying on post-reaction column separation, this novel approach integrates a mild salt formation step followed by a controlled esterification in polar aprotic solvents like DMA or DMF at low temperatures. The true breakthrough lies in the purification strategy, which replaces solid-phase decolorization with the addition of water-soluble reducing agents during liquid-liquid extraction. This subtle yet powerful modification prevents oxidative discoloration at the molecular level, yielding a pristine white crystalline product without the need for filtration aids that trap valuable product. 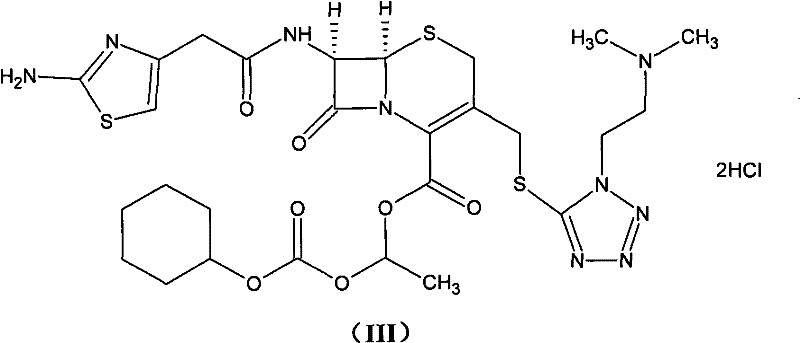
Mechanistic Insights into Bicarbonate-Mediated Esterification
The core of this synthetic advancement relies on the precise generation of the cefotiam salt intermediate using acid bicarbonates such as potassium bicarbonate (KHCO3) or sodium bicarbonate (NaHCO3). Unlike stronger bases that might induce degradation or unwanted side reactions on the sensitive beta-lactam ring, acid bicarbonates provide a buffered environment that facilitates the deprotonation of the carboxylic acid group on the cefotiam nucleus without compromising structural integrity. This salt formation is conducted in a mixed solvent system of water and acetone, which optimizes solubility dynamics to ensure complete conversion while maintaining the stability of the reactive species prior to the esterification step.
Following salt formation, the reaction with 1-iodoethyl cyclohexyl carbonate proceeds under strictly controlled thermal conditions, typically between -10°C and -5°C. This low-temperature regime is critical for kinetic control, suppressing the thermodynamic drive towards isomerization that leads to the undesired Δ2 byproduct. The subsequent workup involves a sophisticated extraction protocol where a water-soluble reducing agent, such as sodium bisulfite or thiosulfate, is introduced directly into the organic solvent layer. This agent scavenges free radicals or oxidizing impurities responsible for color formation, ensuring that the final cefotiam hexetil hydrochloride meets the highest visual and chemical purity standards required for oral antibiotic formulations.
How to Synthesize Cefotiam Hexetil Hydrochloride Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for transitioning from laboratory scale to commercial production. The process begins with the preparation of the key alkylating agent, 1-iodoethyl cyclohexyl carbonate, followed by the in-situ generation of the cefotiam salt. These precursors are then coupled under anhydrous conditions to form the ester, which is subsequently converted to the hydrochloride salt through acidification and recrystallization. The detailed标准化 synthesis steps见下方的指南 ensure that operators can replicate the high yields and purity profiles demonstrated in the patent embodiments.
- React cefotiam with acid bicarbonate (e.g., KHCO3) in a water-acetone mixed solvent to form the cefotiam salt.
- Perform esterification by reacting the cefotiam salt with 1-iodoethyl cyclohexyl carbonate in DMF or DMA at low temperatures (-10 to -5°C).
- Purify the crude ester using ethyl acetate extraction with a water-soluble reducing agent to prevent discoloration, followed by acidification and recrystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, the adoption of this patented process offers distinct strategic advantages over legacy manufacturing methods. By eliminating the need for column chromatography and reducing the number of unit operations required for decolorization, the overall processing time is significantly compressed. This efficiency gain translates directly into higher throughput capacity per reactor volume, allowing manufacturers to meet surging market demand for cephalosporin intermediates without proportional increases in capital expenditure or facility footprint.
- Cost Reduction in Manufacturing: The most immediate financial benefit stems from the dramatic improvement in reaction yield, which consistently exceeds 95% compared to the 20-60% range of older methods. This enhancement means that for every kilogram of starting cefotiam used, nearly double the amount of finished product is recovered, effectively halving the raw material cost per unit of output. Additionally, the removal of expensive purification media like silica gel and activated carbon reduces consumable costs, while the simplified solvent recovery system lowers utility expenses associated with distillation and waste treatment.
- Enhanced Supply Chain Reliability: A simpler process with fewer critical control points inherently reduces the risk of batch failures and production delays. The use of common, commercially available solvents such as ethyl acetate, acetone, and acetonitrile ensures that supply chain disruptions due to specialty chemical shortages are minimized. Furthermore, the robustness of the crystallization step, which tolerates a wider range of operating parameters, ensures consistent batch-to-batch quality, providing procurement managers with the confidence to commit to long-term supply agreements with minimal risk of specification deviations.
- Scalability and Environmental Compliance: The transition from solid-phase decolorization to soluble reducing agents significantly reduces the volume of solid hazardous waste generated during production. This aligns with increasingly stringent environmental regulations and corporate sustainability goals, reducing the burden on waste disposal infrastructure. The process is designed for scalability, utilizing standard extraction and crystallization equipment found in most multipurpose pharmaceutical plants, facilitating a smooth technology transfer from pilot scale to multi-ton commercial production without the need for specialized or custom-engineered reactors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms traditional techniques in terms of purity, yield, and operational simplicity.
Q: How does this method control the isomer content in Cefotiam Hexetil?
A: The method utilizes specific low-temperature esterification conditions (-10 to -5°C) and a controlled molar ratio of reactants, which significantly suppresses the formation of the Δ2 isomer, keeping total impurities below 2%.
Q: What is the role of the water-soluble reducing agent in the purification step?
A: Adding agents like sodium bisulfite or thiosulfate during the extraction phase eliminates color-developing factors, ensuring the final product is a pure white crystal rather than the pink or faint yellow often seen in conventional processes.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process avoids complex column chromatography and uses common industrial solvents like ethyl acetate and acetone, making it highly scalable with yields exceeding 95%.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cefotiam Hexetil Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the theoretical advantages of a patent must be translated into tangible commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high purity specifications and rigorous QC labs required for antibiotic intermediates are met with precision. We understand that delivering a white crystalline product with >98.5% purity is not just a chemical target but a commitment to patient safety and regulatory compliance, and our infrastructure is built to support these demanding standards consistently.
We invite global pharmaceutical partners to collaborate with us on optimizing their cephalosporin supply chains. By leveraging this advanced synthesis technology, we can offer a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments, allowing you to evaluate how our high-efficiency manufacturing capabilities can enhance your product portfolio and reduce time-to-market for your critical antibiotic therapies.