Scalable Synthesis of Remdesivir Intermediates via Column-Free Technology for Commercial Production
Scalable Synthesis of Remdesivir Intermediates via Column-Free Technology for Commercial Production
The global pharmaceutical landscape has witnessed an unprecedented demand for antiviral therapeutics, particularly nucleoside analogs like Remdesivir, following the emergence of novel coronavirus strains. In response to the critical need for efficient manufacturing routes, patent CN111233870A introduces a groundbreaking methodology for the rapid preparation of key Remdesivir pharmaceutical intermediates, specifically the halogenated pyrrolo[2,1-f][1,2,4]triazine derivatives designated as Formula (I). This intellectual property represents a significant leap forward in process chemistry, addressing the bottlenecks of traditional synthesis which often rely on laborious purification techniques. By leveraging a unique four-step continuous sequence that operates primarily in aqueous media during the initial stages, this technology enables the production of high-purity intermediates without the necessity for complex column chromatography. For R&D directors and procurement specialists alike, this patent outlines a pathway that not only simplifies the synthetic workflow but also drastically reduces the environmental footprint and operational costs associated with large-scale API intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of triazineamine derivatives, which serve as the core scaffold for Remdesivir and related nucleoside drugs, has been plagued by inefficiencies that hinder mass production. Traditional routes, such as those described in earlier literature by researchers like Robert S. Klein, typically involve multi-step sequences where the isolation and purification of intermediates require extensive column chromatography. This reliance on silica gel separation is not only time-consuming and solvent-intensive but also presents significant challenges when attempting to scale up from gram-scale laboratory experiments to kilogram or ton-level commercial production. The use of column chromatography introduces variability in yield, increases the risk of product degradation due to prolonged exposure to stationary phases, and generates substantial amounts of hazardous chemical waste. Furthermore, the cumulative yield losses across multiple purification steps often result in overall process economics that are unsustainable for meeting the urgent demands of a global health crisis, making the search for alternative, streamlined methodologies a top priority for the industry.
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in patent CN111233870A offers a robust and streamlined alternative that fundamentally reimagines the purification strategy. The core innovation lies in the ability to drive the reaction sequence to completion with such high selectivity that the final products can be isolated through simple physical means, specifically filtration and recrystallization, rather than chromatographic separation. The process initiates with the conversion of a pyrrole aldehyde to a nitrile using sulfamic acid in water, followed by a direct basification to generate the cyanamide species. This aqueous compatibility in the early stages minimizes the use of volatile organic compounds (VOCs) and allows for straightforward workup procedures. By eliminating the column chromatography bottleneck, this novel approach ensures a more consistent impurity profile, higher throughput, and significantly reduced processing time, thereby providing a reliable foundation for the cost reduction in pharmaceutical intermediates manufacturing that supply chain leaders desperately seek.
Mechanistic Insights into Aqueous-Promoted Cyclization and Halogenation
The chemical elegance of this synthesis is rooted in its strategic use of reagents that facilitate transformation while maintaining product integrity. The initial step involves the reaction of pyrrole-2-carbaldehyde (Formula II) with sulfamic acid in an aqueous solvent. Sulfamic acid acts as a mild yet effective reagent to convert the aldehyde functionality into a nitrile group (Formula III), a transformation that proceeds efficiently at ambient temperatures ranging from 0°C to 25°C. Following this, the addition of a strong base, such as potassium hydroxide or sodium carbonate, directly to the reaction mixture induces the formation of the cyanamide intermediate (Formula VI). This one-pot progression from aldehyde to cyanamide avoids the isolation of unstable intermediates, reducing material loss. The subsequent cyclization step is critical, where the cyanamide reacts with formamidine acetate in an organic solvent like ethanol or methanol at elevated temperatures between 60°C and 100°C. This thermal energy drives the condensation reaction to form the fused pyrrolo-triazine ring system (Formula V), creating the heterocyclic core essential for biological activity.
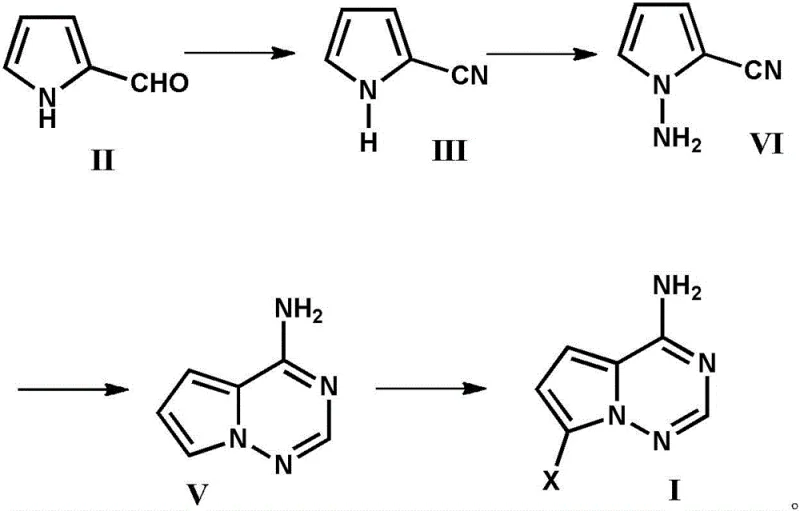
The final stage of the synthesis involves the regioselective halogenation of the triazineamine core to yield the target intermediate (Formula I), where X represents a halogen atom such as iodine, bromine, or chlorine. This is achieved using N-halosuccinimides, such as N-iodosuccinimide (NIS), under mild conditions (0°C to 25°C) in solvents like DMF or dichloromethane. The mechanistic precision of this halogenation ensures that the substitution occurs exclusively at the desired position on the pyrrole ring, minimizing the formation of regioisomers that would otherwise complicate downstream coupling reactions. The ability to achieve such high regioselectivity without the need for chromatographic cleanup is a testament to the optimized reaction conditions described in the patent. This mechanistic robustness is crucial for R&D teams focused on impurity control, as it guarantees a cleaner crude product that meets stringent purity specifications required for GMP manufacturing of antiviral APIs.
How to Synthesize Halogenated Triazineamine Efficiently
Implementing this synthesis route in a production environment requires careful attention to the specific parameters outlined in the patent to ensure reproducibility and safety. The process is designed as a continuous sequence where the output of one step feeds directly into the next, minimizing handling and exposure. Operators must strictly control the temperature profiles, particularly maintaining the low-temperature range during the exothermic basification and halogenation steps to prevent side reactions. The transition from aqueous to organic solvent systems in the third step requires efficient phase management or solvent swapping techniques to optimize the cyclization yield. While the general workflow is simplified, adherence to the specified molar ratios—such as using a significant excess of sulfamic acid in the first step—is vital for driving the reactions to completion. The detailed standardized synthetic steps see the guide below for precise operational protocols.
- React pyrrole-2-carbaldehyde with excess sulfamic acid in water at 0-25°C to form the nitrile compound.
- Adjust the reaction mixture to alkaline pH using a base to convert the nitrile into the cyanamide derivative.
- Condense the cyanamide with formamidine acetate in an organic solvent at 60-100°C to cyclize into the triazineamine core.
- Perform electrophilic halogenation using N-halosuccinimide at 0-25°C to yield the final halogenated intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this column-free synthesis technology translates into tangible operational benefits that extend far beyond the laboratory bench. The primary advantage lies in the drastic simplification of the downstream processing unit operations. By removing the requirement for column chromatography, manufacturers can eliminate the capital expenditure associated with large-scale chromatography columns and the recurring costs of silica gel and vast quantities of elution solvents. This reduction in material consumption directly correlates to a lower cost of goods sold (COGS) and a smaller environmental footprint, aligning with modern green chemistry initiatives. Furthermore, the reliance on simple filtration and drying significantly shortens the batch cycle time, allowing facilities to increase their production capacity and respond more agilely to market fluctuations. This enhanced throughput capability is essential for securing a stable supply of critical antiviral intermediates during periods of high global demand.
- Cost Reduction in Manufacturing: The elimination of column chromatography serves as a major driver for cost optimization in the production of high-purity Remdesivir intermediates. Traditional purification methods are notoriously expensive due to the high volume of solvents required for elution and the cost of disposable stationary phases. By replacing these with filtration and recrystallization, the process significantly reduces solvent recovery loads and waste disposal fees. Additionally, the use of inexpensive and readily available reagents like sulfamic acid and formamidine acetate further lowers the raw material costs. This economic efficiency allows suppliers to offer more competitive pricing structures without compromising on the quality or purity of the final chemical entity, providing a distinct financial advantage in the procurement of complex pharmaceutical building blocks.
- Enhanced Supply Chain Reliability: Supply chain resilience is heavily dependent on the simplicity and robustness of the manufacturing process. Complex purification steps often introduce points of failure, such as column channeling or inconsistent fraction collection, which can lead to batch failures and delivery delays. The streamlined nature of this patented method mitigates these risks by utilizing unit operations that are easily scalable and controllable, such as stirring, heating, and filtration. The ability to perform the initial steps in water also reduces dependency on specialized organic solvents that might face supply constraints. Consequently, this reliability ensures consistent lead times and uninterrupted availability of the intermediate, which is critical for downstream API manufacturers who operate on tight production schedules and cannot afford raw material shortages.
- Scalability and Environmental Compliance: Scaling chemical processes from the lab to the plant floor often reveals hidden complexities, but this methodology is inherently designed for expansion. The absence of chromatography removes a significant barrier to scale-up, as industrial-scale columns are difficult to operate efficiently compared to large filtration units. Moreover, the process generates less hazardous waste, particularly by minimizing the use of chlorinated solvents in the early stages and reducing the overall solvent volume. This aligns with increasingly stringent environmental regulations regarding VOC emissions and chemical waste disposal. Facilities adopting this route can achieve higher production volumes while maintaining compliance with environmental standards, ensuring long-term sustainability and reducing the regulatory burden associated with chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and advantages of the method. Understanding these details is essential for technical teams evaluating the integration of this route into their existing manufacturing portfolios. The answers highlight the specific conditions and outcomes that differentiate this approach from conventional practices, ensuring that stakeholders have a comprehensive understanding of the process capabilities.
Q: Does this synthesis method require column chromatography?
A: No, the patented process specifically eliminates the need for column chromatography, relying instead on simple filtration and recrystallization for purification.
Q: What are the reaction conditions for the cyclization step?
A: The cyclization step (Step 3) requires elevated temperatures between 60°C and 100°C in the presence of a base and an organic solvent like ethanol.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the elimination of complex purification steps and the use of aqueous conditions in early stages make this method highly scalable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Remdesivir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a robust and scalable supply chain for antiviral pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and high-quality supply of materials. Our state-of-the-art facilities are equipped to handle the specific requirements of the column-free synthesis described in patent CN111233870A, utilizing our rigorous QC labs to maintain stringent purity specifications for every batch. We understand that the purity of the intermediate directly impacts the efficacy and safety of the final drug product, which is why our quality assurance protocols are designed to exceed industry standards. By partnering with us, you gain access to a supply chain that is not only reliable but also optimized for speed and efficiency.
We invite potential partners to engage with our technical procurement team to discuss how we can tailor our manufacturing capabilities to your specific needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to evaluate the feasibility of this new synthetic route for your portfolio, our experts are ready to assist. We encourage you to request specific COA data and route feasibility assessments to verify our commitment to quality and transparency. Together, we can accelerate the availability of life-saving medications by ensuring a steady flow of high-quality intermediates, leveraging our technical expertise to overcome the challenges of complex chemical synthesis.