Advanced Synthesis of HIV Protease Inhibitor Intermediates for Commercial Scale Production
Advanced Synthesis of HIV Protease Inhibitor Intermediates for Commercial Scale Production
The pharmaceutical industry continuously seeks more efficient pathways for the production of critical antiretroviral agents, particularly those targeting the Human Immunodeficiency Virus (HIV). Patent CN1190966A discloses a groundbreaking preparation process for Compound J, a clinically valuable HIV protease inhibitor, and its key intermediates. This technology represents a significant departure from traditional synthetic routes by introducing an improved alternative synthesis of the 2-4-picolyl-2-piperazine-tert-butyl carboxamide intermediate. By streamlining the molecular construction of this complex scaffold, the patent offers a robust solution for reducing the step count and enhancing the overall economic viability of manufacturing high-purity API intermediates. The innovation lies in the strategic application of multicomponent reactions and optimized coupling strategies that bypass labor-intensive protection and deprotection sequences.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
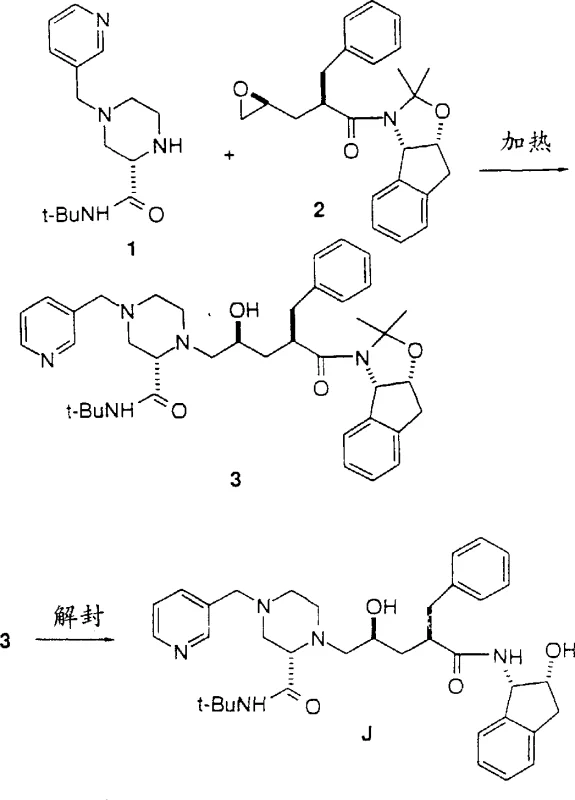
Historically, the synthesis of Compound J and its analogues was achieved through a cumbersome twelve-step procedure, as detailed in earlier literature such as EPO 541,168. In this conventional methodology, the preparation involved coupling a Boc-protected piperazine carboxylic acid amide with an epoxide intermediate to yield a protected coupling product. A major drawback of this legacy route is the necessity for three distinct chemical steps merely to transform the epoxide intermediate into the final compound after the initial coupling. Specifically, the requirement for Boc-protection prior to coupling introduces significant inefficiency, as the protecting group must subsequently be removed in a dedicated deblocking step before a final picolylation can occur. This multi-step sequence not only increases the consumption of raw materials and solvents but also compounds the potential for yield loss at each stage, thereby inflating the cost of goods and extending the production timeline unnecessarily.
The Novel Approach
In stark contrast to the legacy methods, the present invention introduces a highly efficient two-step program for the preparation of HIV protease inhibitor J starting from 4-picolyl piperazine carboxylic acid amides. This novel approach capitalizes on the unexpected reactivity of piperazine 1, which contains three basic amine functional groups capable of attacking the epoxide intermediate directly. Unlike the Boc-protected piperazines which possess only one basic amine functional group, the unprotected or selectively substituted piperazine 1 allows for a high-efficiency condensation with epoxide 2 to provide the coupling product 3 in a single operation. Subsequently, the removal of the acetonide protecting group from intermediate 3 directly yields the final HIV-1 protease inhibitor J. This streamlined pathway effectively eliminates the independent picolylation step required in the conventional route, thereby collapsing the synthesis into fewer operational units and significantly enhancing process throughput.
Mechanistic Insights into Multicomponent Condensation Reactions
The core innovation enabling this streamlined synthesis is the application of advanced multicomponent reaction mechanisms, specifically the Ugi four-component condensation (4CC) and the Strecker synthesis, adapted for piperazine construction. The Ugi reaction is characterized by the alpha-addition of a negatively charged ion and an isocyanide to an iminium ion, followed by a spontaneous rearrangement to form a stable alpha-amino carboxamide derivative. In the context of this patent, the reaction involves the condensation of an imine, a carboxylic acid, an isocyanide, and an amine component to generate the 1-acyl piperazine-2-tert-butyl carboxamide derivative. This one-pot transformation is remarkably efficient as it constructs multiple bonds and stereocenters simultaneously without the need for isolating unstable intermediates. The versatility of this mechanism allows for the use of various carboxylic acids, such as formic acid or benzoic acid, and isocyanides, providing flexibility in tuning the electronic properties of the intermediate.
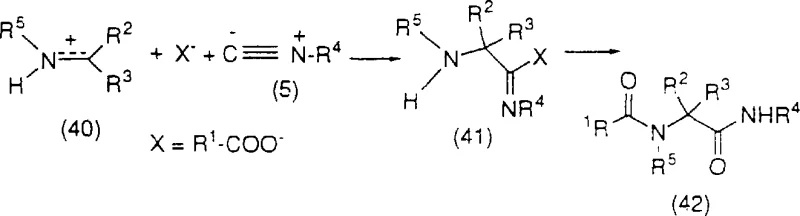
Furthermore, the patent describes a variation involving the Strecker synthesis coupled with a Ritter reaction to achieve similar structural outcomes. In this pathway, an intermediate imine reacts with a cyanide ion to form an amino-nitrile, which is subsequently converted into the desired amide via acid-catalyzed reaction with tert-butanol or isobutylene. This mechanistic diversity provides process chemists with multiple viable routes to access the critical piperazine-2-carboxamide intermediate depending on reagent availability and cost considerations. The ability to perform these transformations in a one-pot manner, without isolating the imine intermediate, drastically reduces solvent waste and handling time. Moreover, the reaction conditions are relatively mild, typically proceeding at temperatures between 0°C and 80°C in solvents such as methanol or ethanol, which are favorable for large-scale operations due to their low cost and ease of removal.
How to Synthesize Piperazine-tert-butyl Carboxamide Efficiently
The synthesis of the key piperazine intermediate is achieved through a carefully controlled sequence of condensation and cyclization reactions. The process begins with the formation of an imine from ethylenediamine derivatives and haloacetaldehyde, followed immediately by the addition of isocyanide and carboxylic acid components for the Ugi variant, or cyanide for the Strecker variant. The reaction mixture is maintained at optimal temperatures, typically ranging from 25°C to 70°C, to ensure complete conversion while minimizing side reactions. Following the formation of the protected piperazine derivative, a deprotection step is employed to remove the N-acyl group, yielding the free piperazine-2-tert-butyl carboxamide. This intermediate is then ready for the critical coupling step with the epoxide fragment.
- Prepare the chiral piperazine-2-carboxylic acid amide intermediate using a one-pot Ugi four-component condensation or Strecker reaction followed by Ritter amidation.
- Couple the resulting piperazine intermediate with the protected epoxide fragment under heated conditions to form the coupled product.
- Perform a final deprotection step using acidic conditions to remove the acetonide protecting group and yield the final HIV protease inhibitor Compound J.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers substantial strategic advantages rooted in process simplification and resource optimization. The primary benefit stems from the drastic reduction in the number of synthetic steps, which directly correlates to lower operational expenditures and reduced capital tie-up in work-in-progress inventory. By eliminating the need for expensive protecting group chemistry, such as Boc-anhydride, and the associated purification steps, the process achieves a leaner manufacturing profile. This efficiency translates into a more competitive cost structure for the final API intermediate, allowing pharmaceutical companies to better manage their COGS (Cost of Goods Sold) in a price-sensitive market. Additionally, the use of commodity chemicals like formic acid, acetaldehyde, and simple alcohols as solvents ensures a stable and reliable supply chain, mitigating risks associated with specialized reagent shortages.
- Cost Reduction in Manufacturing: The elimination of the Boc-protection and subsequent deprotection steps removes the need for costly reagents and the extensive washing procedures required to remove urea byproducts. This qualitative improvement in atom economy means that less raw material is wasted, and the overall yield per batch is significantly enhanced. Furthermore, the ability to perform the key bond-forming reactions in a one-pot fashion reduces the demand for reactor volume and utility consumption, such as steam for heating and chilled water for cooling, leading to substantial overhead savings.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials, such as ethylenediamine derivatives and simple aldehydes, ensures that the supply chain is robust against disruptions. Unlike processes that depend on chiral pool materials with limited global availability, this synthetic route allows for the generation of chirality through resolution or asymmetric catalysis at a later stage, providing flexibility in sourcing. The simplified workflow also reduces the lead time for production campaigns, enabling faster response to market demand fluctuations and ensuring continuity of supply for critical HIV medications.
- Scalability and Environmental Compliance: The process is inherently scalable due to its use of standard unit operations like heating, stirring, and crystallization, which are easily transferred from pilot plant to commercial scale. The reduction in solvent usage and the avoidance of heavy metal catalysts or hazardous reagents align with modern green chemistry principles, simplifying waste treatment and regulatory compliance. This environmental stewardship not only reduces disposal costs but also enhances the corporate sustainability profile of the manufacturing entity, a key metric for modern pharmaceutical partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and process descriptions provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers. Understanding these nuances is critical for evaluating the feasibility of integrating this route into existing manufacturing frameworks.
Q: How does the new synthesis route improve upon conventional methods for Compound J?
A: The novel process eliminates the need for Boc-protection and subsequent deprotection steps required in conventional 12-step syntheses. By utilizing a direct picolyl-piperazine intermediate, the route reduces the total number of chemical transformations, significantly lowering material costs and processing time while improving overall yield.
Q: What are the key reaction mechanisms utilized in this patented process?
A: The process leverages versatile multicomponent reactions, specifically the Ugi four-component condensation or a modified Strecker synthesis combined with a Ritter reaction. These mechanisms allow for the efficient construction of the complex piperazine core in a one-pot fashion, minimizing isolation steps and solvent usage.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is designed for scalability. It utilizes robust reaction conditions, such as heating in alcohol solvents at 65-85°C, and avoids sensitive reagents that require cryogenic temperatures. The simplification of purification steps, often relying on crystallization rather than complex chromatography, makes it highly viable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Piperazine-tert-butyl Carboxamide Supplier
As a leader in the fine chemical sector, NINGBO INNO PHARMCHEM is uniquely positioned to leverage this advanced synthesis technology for the commercial production of HIV protease inhibitor intermediates. Our facility boasts extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of piperazine-tert-butyl carboxamide meets the highest quality standards required for pharmaceutical applications. Our commitment to technical excellence ensures that the complex stereochemistry and structural integrity of these critical intermediates are preserved throughout the manufacturing process.
We invite global pharmaceutical partners to collaborate with us to optimize their supply chains for antiretroviral therapies. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage potential clients to contact us directly to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions based on empirical evidence and our proven track record in delivering high-value pharmaceutical intermediates.