Advanced Manufacturing Strategies For HIV Protease Inhibitor Intermediates And Commercial Scale-Up
Introduction To Streamlined HIV Protease Inhibitor Synthesis
The pharmaceutical industry continuously seeks robust and efficient pathways for the production of complex antiretroviral agents, particularly those targeting the Human Immunodeficiency Virus (HIV). Patent CN1491945A discloses a significant technological breakthrough in the preparation of Compound J, a potent HIV protease inhibitor, by introducing a novel synthetic route for its critical piperazine-tert-butylcarboxamide intermediates. This innovation addresses long-standing inefficiencies in the prior art, specifically the cumbersome 12-step procedures previously described in EPO 541,168. By leveraging advanced multicomponent condensation reactions such as the Ugi, Strecker, and Ritter reactions, the disclosed process achieves a dramatic simplification of the molecular architecture assembly. This technical advancement is not merely an academic exercise but represents a tangible shift towards more sustainable and economically viable manufacturing protocols for high-value pharmaceutical intermediates. The ability to bypass multiple protection and deprotection cycles while maintaining high stereochemical integrity offers a compelling value proposition for large-scale production facilities aiming to optimize their operational expenditure and reduce environmental footprint.
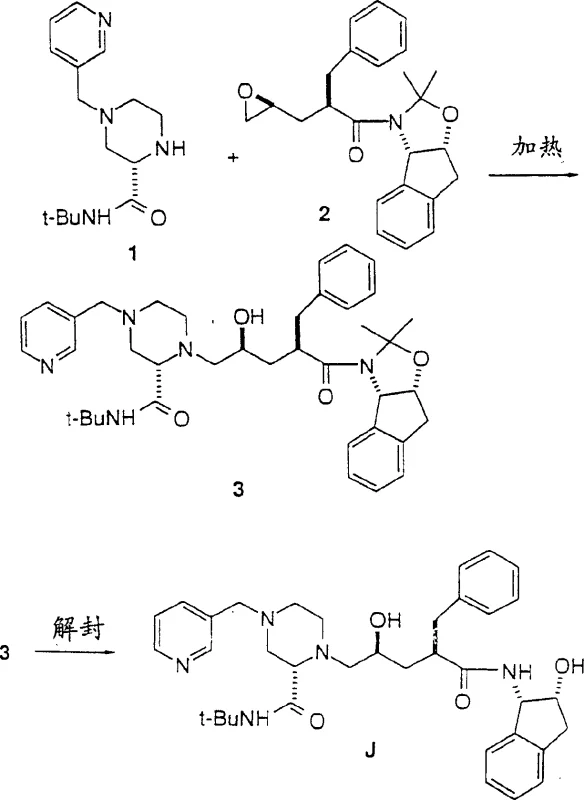
The Limitations Of Conventional Methods Vs. The Novel Approach
The Limitations Of Conventional Methods
Historically, the synthesis of Compound J and its analogs relied on a linear and labor-intensive 12-step sequence that posed significant challenges for industrial scalability. In the conventional methodology, the construction of the key piperazine scaffold required the initial preparation of a pyrazine-2-tert-butylcarboxamide precursor, followed by a hydrogenation step to reduce the aromatic ring to a racemic piperazine. This approach necessitated a subsequent resolution step using optically active acids to isolate the desired enantiomer, adding considerable cost and complexity. Furthermore, the differentiation of the nitrogen atoms at the N1 and N4 positions of the piperazine ring required selective protection strategies, typically involving bulky Boc groups. The introduction of the 3-picolyl moiety at the 4-position was particularly problematic, often resulting in poor selectivity ratios of mono- to dialkylation products, which created difficult-to-purify mixtures. Consequently, the traditional route demanded three distinct chemical steps just to convert the epoxide intermediate into the final inhibitor, including a separate picolylation step after deblocking, which inherently increased the risk of yield loss and impurity generation at every stage of the workflow.
The Novel Approach
In stark contrast, the novel approach detailed in the patent data revolutionizes this landscape by enabling the direct coupling of a pre-functionalized 4-picolylpiperazine carboxamide with the epoxide intermediate. This strategy effectively consolidates the synthesis into a convenient two-step procedure, eliminating the need for the transient Boc protection of the reactive 4-position prior to coupling. Although the presence of three basic amine functional groups in the picolyl-piperazine intermediate initially suggested potential issues with regioselectivity during the epoxide ring-opening, the process demonstrates unexpectedly high efficiency. The condensation of piperazine 1 with epoxide 2 directly yields the coupled product 3, which subsequently undergoes a straightforward removal of the acetonide protecting group to afford the final HIV-1 protease inhibitor J. This reduction in step count not only accelerates the overall throughput but also minimizes the accumulation of by-products and solvent waste, thereby enhancing the overall mass balance of the manufacturing campaign. The elimination of the separate picolylation step post-deblocking is a critical optimization that streamlines the downstream processing and purification requirements.
Mechanistic Insights Into Multicomponent Condensation Reactions
The core of this technological leap lies in the innovative application of multicomponent reactions for the construction of the chiral piperazine-2-carboxamide backbone. The patent elucidates the utility of the Ugi four-component condensation, where an imine intermediate, generated in situ from an ethylenediamine derivative and a halogenated acetaldehyde, reacts with an isocyanide and a carboxylic acid. This mechanism facilitates the simultaneous formation of the piperazine ring and the amide bond in a single pot, bypassing the need for isolating unstable imine intermediates. The reaction proceeds through the alpha-addition of the isocyanide and the carboxylate anion to the iminium ion, followed by a spontaneous acyl transfer rearrangement to yield the stable alpha-aminocarboxamide derivative. Alternatively, the Strecker variant utilizes a cyanide ion to trap the imine, forming an aminonitrile which can be subsequently converted to the amide via a Ritter reaction. These mechanistic pathways offer superior atom economy compared to the traditional stepwise assembly from aromatic pyrazine precursors. The flexibility to utilize various carboxylic acids, such as benzoic acid or formic acid, allows for fine-tuning the electronic properties of the intermediate, while the use of tert-butyl isocyanate ensures the installation of the requisite tert-butylcarboxamide motif with high fidelity.
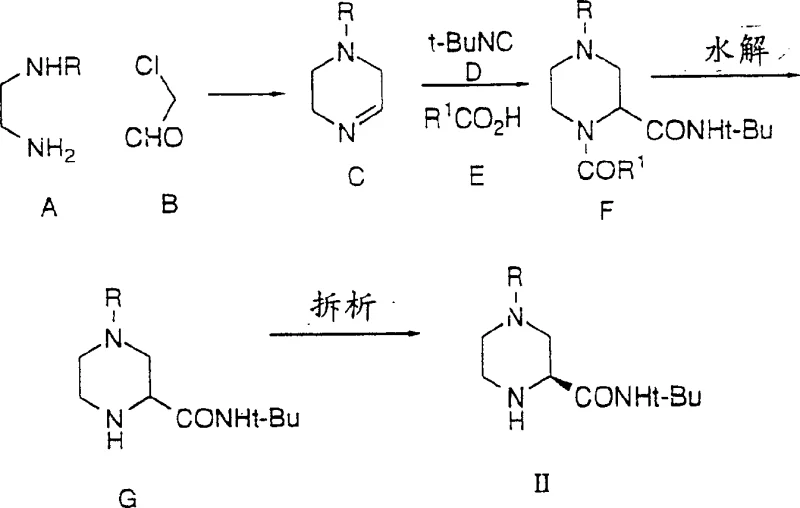
Furthermore, the control of stereochemistry and impurity profiles is meticulously managed through the selection of reaction conditions and resolving agents. The process acknowledges the formation of racemic mixtures during the initial ring closure and provides robust methods for chiral resolution, such as salt formation with (S)-camphorsulfonic acid or L-pyroglutamic acid. This ensures that only the biologically active (S)-enantiomer proceeds to the final coupling stage. The mechanistic understanding extends to the management of side reactions; for instance, the potential for over-alkylation or hydrolysis of sensitive functional groups is mitigated by precise temperature control, typically maintaining reaction environments between 0°C and 80°C depending on the specific transformation. The use of strong acids like sulfuric acid in the Ritter reaction variant is carefully balanced to promote the conversion of nitriles to amides without compromising the integrity of the piperazine ring. This deep mechanistic insight allows process chemists to predict and prevent the formation of genotoxic impurities or difficult-to-remove by-products, ensuring that the final intermediate meets the stringent purity specifications required for clinical-grade pharmaceutical manufacturing.
How To Synthesize Compound J Efficiently
The practical execution of this synthesis involves a sequence of highly optimized unit operations designed to maximize yield and minimize operational hazards. The process begins with the preparation of the key piperazine intermediate, where ethylenediamine derivatives are condensed with chloroacetaldehyde in the presence of a cyanide source or isocyanide, depending on the chosen pathway. Following the isolation and resolution of the chiral piperazine carboxamide, the critical coupling step is performed by heating the amine with the epoxide partner in a suitable solvent system such as tert-amyl alcohol or isopropanol. The reaction mixture is then subjected to acidic hydrolysis to remove the acetonide protecting group, revealing the free hydroxyl groups essential for biological activity. Detailed standardized synthetic steps for this procedure are provided in the guide below to ensure reproducibility and safety compliance.
- Condense 4-picolylpiperazine carboxamide with the epoxide intermediate under heated conditions to form the coupled product.
- Perform deblocking of the acetonide protecting group using strong acid treatment to yield the final HIV protease inhibitor Compound J.
Commercial Advantages For Procurement And Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route translates into substantial strategic advantages regarding cost structure and supply reliability. The reduction in the total number of synthetic steps directly correlates with a decrease in the consumption of raw materials, solvents, and reagents, leading to a lower cost of goods sold (COGS). By eliminating the need for expensive protecting group reagents like di-tert-butyl dicarbonate and the associated scavengers for their removal, the process achieves significant cost reduction in pharmaceutical manufacturing without compromising quality. Furthermore, the simplification of the workflow reduces the demand for reactor occupancy time, allowing facilities to increase their production throughput and respond more agilely to market demands. The avoidance of complex purification sequences, such as those required to separate mono- and di-alkylated by-products in the old method, enhances the overall equipment effectiveness and reduces the burden on waste treatment systems. This streamlined approach ensures a more robust supply chain capable of sustaining continuous commercial production with fewer interruptions due to process upsets or yield fluctuations.
- Cost Reduction In Manufacturing: The elimination of multiple protection and deprotection cycles removes the need for costly reagents and the extensive washing procedures associated with them. This qualitative improvement in process efficiency leads to substantial savings in utility consumption and waste disposal costs, as fewer solvent exchanges and distillation steps are required to isolate the intermediates. The one-pot nature of the Ugi and Strecker variants further consolidates operations, reducing labor hours and minimizing the risk of material loss during transfers between vessels.
- Enhanced Supply Chain Reliability: By utilizing readily available starting materials such as ethylenediamine, chloroacetaldehyde, and simple carboxylic acids, the process mitigates the risk of supply bottlenecks associated with specialized or custom-synthesized precursors. The robustness of the reaction conditions, which tolerate a range of temperatures and solvent systems, ensures consistent batch-to-batch quality, thereby reducing the likelihood of failed batches that could disrupt the supply of the final active pharmaceutical ingredient. This reliability is crucial for maintaining uninterrupted therapy for patients dependent on HIV medications.
- Scalability And Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that are easily transferable from laboratory to pilot and commercial scales. The reduction in waste by-product formation aligns with green chemistry principles, facilitating easier compliance with increasingly stringent environmental regulations. The ability to crystallize intermediates directly from the reaction mixture simplifies isolation and reduces the volume of organic waste streams, contributing to a more sustainable manufacturing footprint that appeals to environmentally conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis pathway. These insights are derived directly from the experimental data and embodiments described in the patent documentation, providing clarity on critical process parameters and troubleshooting strategies. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this technology for their own manufacturing portfolios.
Q: How does the new process improve upon the conventional 12-step synthesis?
A: The novel process eliminates the need for Boc protection and separate picolylation steps, reducing the synthesis from three chemical steps to just two direct transformations, significantly lowering material costs and processing time.
Q: What are the key advantages of using Ugi or Strecker reactions for the piperazine intermediate?
A: These multicomponent reactions allow for the one-pot formation of the piperazine ring and amide bond from simple starting materials like ethylenediamine and chloroacetaldehyde, avoiding the labor-intensive reduction of aromatic pyrazine precursors.
Q: Is chiral resolution required for the intermediates?
A: Yes, separation of (S) and (R) enantiomers is necessary, typically achieved via chiral HPLC or salt formation with resolving agents like camphorsulfonic acid or tartaric acid to ensure the desired stereochemistry for biological activity.
Partnering With NINGBO INNO PHARMCHEM: Your Reliable Compound J Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable processes in the production of life-saving antiretroviral therapies. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless and secure. We are committed to delivering high-purity HIV protease inhibitor intermediates that meet rigorous quality standards, supported by our state-of-the-art rigorous QC labs and comprehensive analytical capabilities. Our facility is equipped to handle complex multicomponent reactions and chiral resolutions with precision, guaranteeing the stereochemical integrity required for therapeutic efficacy.
We invite global pharmaceutical partners to collaborate with us to leverage these advanced manufacturing technologies for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality specifications. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that demonstrate how our optimized processes can enhance your operational efficiency and reduce time-to-market for your critical drug products.