Revolutionizing Etoposide Manufacturing: A Streamlined 3-Step Synthetic Route for Commercial Scale
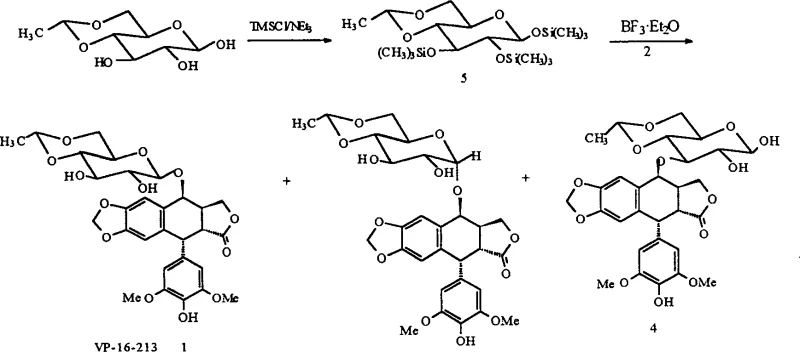
The pharmaceutical landscape for antineoplastic agents continues to evolve, driven by the urgent need for more efficient and cost-effective manufacturing processes for critical drugs like Etoposide (VP-16). Patent CN1172945C introduces a groundbreaking synthetic methodology that fundamentally alters the production economics of this essential chemotherapy agent. By leveraging a stereospecific silylation strategy followed by Lewis acid-catalyzed glycosylation, this technology reduces the synthetic complexity from traditional multi-step sequences to a concise three-step pathway starting from D-glucose derivatives. For R&D directors and procurement specialists alike, this represents a significant opportunity to optimize the supply chain for high-purity pharmaceutical intermediates. The core innovation lies in the direct conversion of 4,6-O,O-ethylene-D-glucose into a fully silylated beta-configured intermediate, which then couples directly with epipodophyllotoxin. This approach not only enhances the overall yield but also mitigates the risks associated with lengthy synthetic routes, such as cumulative yield loss and impurity accumulation. As a reliable etoposide intermediate supplier, understanding these mechanistic advantages is crucial for ensuring consistent quality and supply continuity in the global oncology market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Etoposide has been plagued by inefficiencies inherent in classical glycosylation strategies. Conventional routes, such as those developed by Sandoz or the Tokyo Institute of Microbial Chemistry, typically involve eight or more distinct chemical transformations starting from glucose. These legacy methods often rely on the Koenigs-Knorr reaction mechanism, which necessitates the preparation of glycosyl halides and the use of expensive silver salts like silver carbonate to promote the coupling reaction. Furthermore, achieving the critical beta-stereochemistry at the anomeric center often requires elaborate protecting group manipulations at the C-2 position of the sugar moiety to direct the stereochemical outcome. This results in a process that is not only capital intensive due to the high cost of reagents and solvents but also operationally complex, requiring rigorous control over multiple isolation and purification stages. The cumulative effect of these additional steps is a significantly lower overall yield and a larger environmental footprint due to increased solvent consumption and waste generation, posing challenges for cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
In stark contrast, the methodology disclosed in CN1172945C offers a streamlined alternative that bypasses many of these historical bottlenecks. The novel approach utilizes trimethylchlorosilane (TMSCl) to simultaneously protect the hydroxyl groups at positions 1, 2, and 3 of the 4,6-O,O-ethylene-D-glucose scaffold. This single transformation stereospecifically yields the 1,2,3-O-tri-trimethylsilyl-beta-D-glucose derivative with an impressive yield of 93%. This silylated intermediate serves as a highly reactive glycosyl donor that can couple directly with epipodophyllotoxin under mild Lewis acid catalysis. By eliminating the need for halogenation and silver-promoted activation, the process drastically simplifies the workflow. The reduction from an eight-step sequence to a mere three steps from the ethylidene glucose precursor translates directly into enhanced process robustness. This efficiency gain is pivotal for the commercial scale-up of complex pharmaceutical intermediates, allowing manufacturers to achieve higher throughput with reduced operational overhead and a smaller physical plant footprint.
Mechanistic Insights into Lewis Acid-Catalyzed Stereoselective Glycosylation
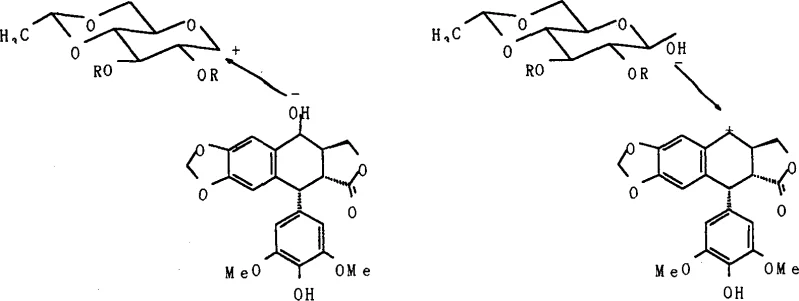
The success of this synthetic route hinges on a deep understanding of the stereoelectronic effects governing the glycosylation event. The reaction mechanism likely proceeds via a pathway distinct from the traditional SN1-type displacement seen in halide-based methods. In the presence of a Lewis acid such as boron trifluoride ether complex (BF3·Et2O), the trimethylsilyl group at the anomeric position acts as an excellent leaving group, generating an oxocarbenium ion intermediate. However, unlike the non-stereoselective nature of some SN1 processes, the specific conformation of the 4,6-O,O-ethylene protected ring and the neighboring group effects facilitate a highly selective attack. The background literature suggests this may align with a Kuhn-type reaction mechanism, where the nucleophilic attack by the 9-hydroxyl group of epipodophyllotoxin occurs preferentially from the less hindered beta-face. This is corroborated by the spectroscopic data provided in the patent, where the coupling constant J1,2 is observed at 7.3 Hz. This value falls squarely within the range expected for a diaxial relationship (Ja,a = 7-10 Hz), confirming the formation of the desired beta-glycosidic linkage with high optical purity.
Furthermore, the choice of reagents plays a critical role in impurity control and process safety. The use of BF3·Et2O as a catalyst offers a distinct advantage over heavy metal promoters by avoiding the introduction of toxic metal residues that require stringent removal protocols to meet ICH Q3D guidelines for elemental impurities. The reaction conditions are maintained at low temperatures, typically between 0°C and -50°C, which helps to suppress side reactions such as the formation of alpha-anomers or orthoesters. Although the patent notes the formation of minor byproducts, including the alpha-condensate and a 3-O-linked isomer, these impurities are effectively managed through standard column chromatography and recrystallization techniques. The ability to generate the active beta-isomer directly without requiring an anomeric equilibration step post-synthesis significantly reduces the burden on downstream purification. This mechanistic clarity provides R&D teams with the confidence to scale the process, knowing that the stereochemical integrity of the final API is built into the fundamental chemistry of the glycosylation step rather than relying on difficult separations.
How to Synthesize Etoposide Efficiently
The implementation of this synthesis protocol requires precise control over reaction parameters to maximize the yield of the beta-anomer while minimizing the formation of regioisomers. The process begins with the preparation of the silylated sugar donor, followed by the critical coupling reaction with the podophyllotoxin aglycone. Detailed operational guidelines regarding stoichiometry, temperature ramps, and workup procedures are essential for reproducibility. The following section outlines the standardized synthesis steps derived from the patent examples, serving as a foundational guide for process chemists aiming to adopt this technology.
- Prepare 4,6-O,O-ethylene-1,2,3-O-tri-trimethylsilane-beta-D-glucose by reacting 4,6-O,O-ethylene-D-glucose with trimethylchlorosilane and triethylamine in dichloromethane at 0 to -50°C.
- Condense the silylated glucose intermediate with epipodophyllotoxin in dichloromethane using boron trifluoride etherate as a Lewis acid catalyst at low temperature.
- Neutralize the reaction mixture with pyridine, perform aqueous workup, and purify the crude product via column chromatography followed by recrystallization to obtain pure Etoposide.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this streamlined synthesis route offers tangible strategic benefits beyond mere technical elegance. The most immediate impact is seen in the reduction of raw material costs and processing time. By condensing the synthetic sequence, the demand for solvents, reagents, and labor hours is substantially decreased. This efficiency translates into a more competitive cost structure for the final active pharmaceutical ingredient, allowing for better margin management in a price-sensitive generic drug market. Moreover, the reliance on commodity chemicals like trimethylchlorosilane and boron trifluoride, rather than specialized silver salts or complex halogenated sugars, enhances supply chain resilience. These reagents are widely available from multiple global suppliers, reducing the risk of single-source bottlenecks that can disrupt production schedules.
- Cost Reduction in Manufacturing: The elimination of multiple protection and deprotection steps inherently lowers the cost of goods sold (COGS). In traditional routes, each additional step incurs costs related to solvent purchase, recovery, and disposal, as well as energy consumption for heating and cooling. By reducing the step count from eight to three, the process significantly minimizes these variable costs. Additionally, the high yield of the silylation step (93%) ensures that the expensive starting materials are utilized with maximum efficiency, reducing waste and improving the overall atom economy of the process. This lean manufacturing approach allows for substantial cost savings without compromising on the quality or purity of the final product.
- Enhanced Supply Chain Reliability: Shorter synthetic routes inherently possess lower operational risk. With fewer unit operations, there are fewer opportunities for equipment failure, human error, or batch rejection. This reliability is crucial for maintaining continuous supply to downstream formulation partners. The use of robust, well-understood chemical transformations further stabilizes the supply chain, as the process is less sensitive to minor variations in raw material quality compared to more fragile multi-step sequences. Consequently, manufacturers can offer more reliable lead times and guarantee supply continuity even during periods of high market demand or raw material volatility.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method offers a greener profile. The avoidance of silver salts eliminates the generation of heavy metal waste streams, which are costly and difficult to treat. The reduced solvent usage per kilogram of product aligns with modern sustainability goals and regulatory pressures to minimize the environmental footprint of pharmaceutical manufacturing. The process is designed to be scalable, moving seamlessly from laboratory bench scale to multi-ton commercial production. This scalability ensures that the technology can meet the growing global demand for Etoposide while adhering to increasingly stringent environmental regulations regarding waste discharge and solvent emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and mechanistic descriptions provided in the patent documentation, offering clarity on process feasibility and product quality.
Q: What is the primary advantage of the silylation method for Etoposide synthesis?
A: The primary advantage is the significant reduction in synthetic steps. Traditional routes often require up to 8 steps from glucose involving complex protecting group strategies. This patented method achieves the key beta-glycoside intermediate in just two steps from the ethylidene glucose precursor with a high yield of 93%, drastically simplifying the process.
Q: How is stereochemical purity ensured in this synthesis?
A: Stereochemical purity is ensured through the stereospecific silylation of the glucose derivative. The resulting 1,2,3-O-tri-trimethylsilyl intermediate exhibits a J1,2 coupling constant of 7.3 Hz in 1H NMR, which confirms the exclusive formation of the beta-configuration required for biological activity, avoiding the formation of inactive alpha-isomers common in other methods.
Q: Does this method avoid the use of expensive heavy metal catalysts?
A: Yes, unlike some conventional Koenigs-Knorr type reactions that may utilize silver salts (such as Ag2CO3) for halide abstraction, this method utilizes boron trifluoride ether complex (BF3·Et2O) as the Lewis acid catalyst. This substitution eliminates the need for costly heavy metals and simplifies the downstream purification and waste treatment processes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Etoposide Supplier
The technological advancements detailed in CN1172945C underscore the potential for significant optimization in the production of antineoplastic agents. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate such innovative synthetic routes into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the benefits of this streamlined chemistry are realized at an industrial level. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Etoposide intermediate meets the highest international standards for safety and efficacy. Our commitment to quality assurance ensures that the stereochemical integrity and impurity profiles of our products are consistently monitored and controlled.
We invite pharmaceutical partners to collaborate with us to leverage this efficient synthesis method for their supply chains. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments. Together, we can drive down costs and enhance the reliability of the global supply for this critical cancer therapy, ensuring that patients have timely access to life-saving medications.