Advanced Synthesis of Doripenem Intermediates via Novel Pyrrolidine Derivatives for Commercial Scale-up
Advanced Synthesis of Doripenem Intermediates via Novel Pyrrolidine Derivatives for Commercial Scale-up
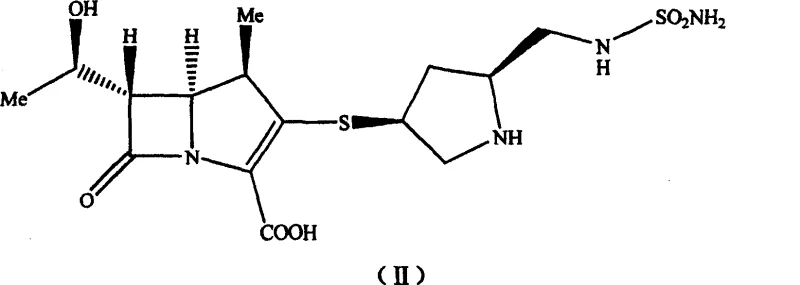
The pharmaceutical industry continuously seeks robust synthetic pathways for next-generation carbapenem antibiotics, particularly for critical agents like Doripenem (S-4661). A pivotal advancement in this domain is detailed in patent CN100460389C, which discloses a novel pyrrolidine derivative intermediate and its application in the preparation of 1β-methyl carbapenem antibiotics. This technology addresses long-standing bottlenecks associated with traditional protecting group strategies, specifically eliminating the reliance on tert-butoxycarbonyl (Boc) groups that necessitate harsh acidolytic cleavage. By shifting to a p-nitrobenzyloxycarbonyl (PNZ) protection strategy, the process not only streamlines the reaction sequence but also fundamentally alters the physical properties of key intermediates, transforming them from difficult-to-handle oils into manageable solids. For R&D directors and process chemists, this represents a significant leap forward in impurity profile control, while supply chain managers will recognize the inherent value in a route that avoids expensive metal scavengers and complex chromatographic purifications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial production of S-4661 has been plagued by two primary synthetic routes, both of which suffer from severe scalability and purity drawbacks that impact cost and yield. The first conventional route utilizes a benzhydryl ester intermediate which is not only costly and scarce in domestic supply chains but also requires the use of Lewis acid aluminum chloride for deprotection. This step introduces a critical contamination risk, as residual aluminum ions are notoriously difficult to remove from the final product, often necessitating the use of divinylbenzene-type macroporous resin columns for purification. The second conventional route attempts to mitigate some costs by using sulfuric acid for tert-butoxycarbonyl removal; however, this acidic environment promotes the formation of reactive carbocations. These carbocations lead to unpredictable side reactions and polymeric impurities that are extremely challenging to separate. Furthermore, the intermediate generated in this acidolysis step exists as an oil at room temperature, creating significant difficulties for accurate metering and feeding in large-scale industrial reactors, thereby compromising batch-to-batch consistency.
The Novel Approach
In stark contrast to these legacy methods, the novel approach described in the patent utilizes a pyrrolidine derivative intermediate that completely eschews the Boc protecting group in favor of the PNZ group. This strategic modification allows for a deprotection sequence that avoids acidolysis entirely, thereby eliminating the generation of carbocation-related impurities and the associated purification nightmares. A crucial advantage of this new intermediate is its physical state; upon deacetylation, the resulting mercaptopyrrolidine derivative isolates as a pressed powder rather than an oil. This physical transformation is vital for industrial operations, as it enables precise gravimetric feeding and simplifies handling protocols. By circumventing the need for expensive benzhydryl esters and toxic Lewis acids, this route offers a cleaner, more direct path to the final antibiotic, ensuring higher purity profiles and reduced operational complexity.
Mechanistic Insights into PNZ-Protected Pyrrolidine Coupling
The core of this technological breakthrough lies in the efficient construction of the sulfamoyl-substituted pyrrolidine side chain using a modified Mitsunobu-type coupling strategy. The synthesis begins with the preparation of N-p-nitrobenzyloxycarbonyl (PNZ) sulfonamide, achieved by reacting p-nitrobenzyl alcohol with chloro sulfonyl isocyanate at cryogenic temperatures (-40°C) followed by ammonolysis. This sulfonamide is then coupled with (2S,4S)-4-acetylthio-1-PNZ-pyrrolidine-2-methanol using triphenylphosphine and diisopropyl azodicarboxylate (DIAD) as the condensing system. The reaction proceeds through the formation of an active alkoxyphosphonium intermediate, which facilitates the nucleophilic attack by the sulfonamide nitrogen. This mechanism is highly stereoselective, preserving the critical (2S,4S) configuration required for biological activity, and proceeds with high efficiency, yielding the protected intermediate in up to 88% yield after recrystallization. The use of PNZ here is mechanistically superior because it remains stable during the coupling conditions but can be cleanly removed later via hydrogenolysis, unlike acid-labile groups.
Following the coupling, the acetylthio protecting group is removed via hydrolysis using mineral alkalis such as lithium hydroxide or sodium hydroxide in a tetrahydrofuran-water system. This deacetylation step is critical for generating the free thiol necessary for the subsequent nucleophilic substitution onto the carbapenem core. Unlike the oily intermediates produced in sulfuric acid deprotection routes, the alkali-treated product precipitates as a solid upon acidification and solvent manipulation. This solid-state isolation acts as a built-in purification step, effectively washing away soluble impurities and unreacted starting materials. The resulting mercaptopyrrolidine derivative is then reacted with the carbapenem phosphonate core in DMF using a tertiary amine base. The nucleophilic attack of the thiol on the activated carbapenem ring proceeds smoothly at ice-bath temperatures, minimizing the risk of ring-opening degradation. The final deprotection via catalytic hydrogenolysis removes both PNZ groups simultaneously, delivering the final S-4661 API with a significantly cleaner impurity profile compared to aluminum chloride-mediated routes.
How to Synthesize Doripenem Intermediate Efficiently
The synthesis of this high-value intermediate requires precise control over reaction parameters, particularly temperature and stoichiometry, to ensure optimal yield and stereochemical integrity. The process outlined in the patent provides a robust framework for laboratory and pilot-scale production, emphasizing the importance of cryogenic conditions during the initial sulfonamide formation and careful pH control during the isolation of the thiol intermediate. Operators must adhere strictly to the specified molar ratios, particularly the 1:1 to 1.5 ratio between the carbapenem core and the pyrrolidine thiol, to maximize conversion while minimizing waste. The following guide summarizes the critical operational phases derived from the patented embodiments, serving as a foundational reference for process implementation.
- Preparation of N-p-nitrobenzyloxycarbonyl (PNZ) sulfonamide by reacting p-nitrobenzyl alcohol with chloro sulfonyl isocyanate followed by ammonolysis and recrystallization.
- Coupling of the sulfonamide with (2S,4S)-4-acetylthio-1-PNZ-pyrrolidine-2-methanol using triphenylphosphine and diisopropyl azodicarboxylate (DIAD) to form the protected intermediate.
- Deacetylation of the coupled product using aqueous alkali (LiOH or NaOH) to yield the free mercaptopyrrolidine derivative ready for carbapenem coupling.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route translates directly into enhanced operational stability and significant cost optimization opportunities. The elimination of specialized reagents such as Lewis acid aluminum chloride and the avoidance of macroporous resin columns for purification drastically simplify the material supply chain. Traditional routes often require sourcing high-purity benzhydryl esters, which can be subject to market volatility and supply constraints; this new method utilizes more commoditized starting materials like p-nitrobenzyl alcohol and chloro sulfonyl isocyanate. Furthermore, the transition from oily intermediates to solid powders reduces the risk of handling errors and spillage during manufacturing, leading to better overall mass balance and yield accountability. These factors collectively contribute to a more resilient supply chain capable of meeting the rigorous demands of global pharmaceutical production.
- Cost Reduction in Manufacturing: The most immediate financial benefit arises from the removal of expensive and hazardous purification steps. By avoiding the use of aluminum chloride, manufacturers eliminate the downstream costs associated with metal scavenging and the disposal of aluminum-contaminated waste streams. Additionally, the ability to isolate intermediates via simple filtration and crystallization, rather than energy-intensive column chromatography, significantly lowers utility consumption and solvent usage. The high yields reported in the patent embodiments, such as the 98.5% yield in the nucleophilic substitution step, further drive down the cost of goods sold by maximizing the output from each batch of raw materials.
- Enhanced Supply Chain Reliability: The physical nature of the intermediates plays a crucial role in supply chain reliability. Oily intermediates, common in older routes, are prone to degradation and are difficult to store or transport safely. In contrast, the pressed powder form of the mercaptopyrrolidine derivative ensures long-term stability and ease of inventory management. This stability allows for the decoupling of synthesis steps, enabling manufacturers to stockpile key intermediates without fear of quality degradation, thus buffering against upstream supply disruptions. The simplified workflow also reduces the dependency on highly specialized contract manufacturing organizations that possess specific resin-column capabilities, broadening the pool of potential suppliers.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this route offers substantial advantages. The avoidance of heavy metals like aluminum aligns with increasingly stringent ICH Q3D guidelines regarding elemental impurities in drug products. Reducing the metal load at the source minimizes the regulatory burden during the filing process and reduces the risk of batch rejection due to out-of-specification metal content. Moreover, the streamlined workup procedures generate less hazardous waste, supporting corporate sustainability goals. The process is inherently scalable, as the unit operations involved—cooling, filtration, and crystallization—are standard in multipurpose chemical plants, facilitating a smooth transition from kilogram-scale development to multi-ton commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this pyrrolidine derivative technology. These insights are derived directly from the comparative data and experimental embodiments provided in the patent literature, offering clarity on why this method is becoming the preferred choice for modern carbapenem manufacturing. Understanding these nuances is essential for stakeholders evaluating the feasibility of technology transfer or process optimization projects.
Q: Why is the PNZ protecting group superior to Boc for Doripenem synthesis?
A: The PNZ (p-nitrobenzyloxycarbonyl) group avoids the harsh acidolysis conditions required for Boc removal. Traditional Boc deprotection often generates carbocation side reactions and oily intermediates that are difficult to purify, whereas the PNZ route yields solid intermediates amenable to simple filtration and crystallization.
Q: How does this process improve impurity control compared to Route 1?
A: Route 1 relies on Lewis acid aluminum chloride for deprotection, which leaves residual aluminum ions that are notoriously difficult to eliminate from the final API. The novel method utilizes hydrogenolysis for final deprotection, completely bypassing the need for metal scavengers or macroporous resin columns.
Q: Is this synthetic route suitable for large-scale manufacturing?
A: Yes, the process is highly scalable because the key mercaptopyrrolidine intermediate isolates as a pressed powder rather than an oil. This physical state significantly facilitates accurate metering and feeding in industrial reactors, solving a major bottleneck found in previous sulfuric acid deprotection methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Doripenem Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of complex antibiotics like Doripenem hinges on the reliability and purity of the supply chain. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from benchtop discovery to full-scale manufacturing. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of detecting trace impurities at ppm levels, guaranteeing that every batch of intermediate meets the exacting standards required for GMP API synthesis. We understand the critical nature of carbapenem intermediates and have optimized our processes to deliver consistent quality with minimal lead times.
We invite you to leverage our technical expertise to optimize your own supply chain. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced PNZ-based synthesis can enhance your production efficiency. Let us partner with you to secure a stable, high-quality supply of Doripenem intermediates that drives your business forward.