Advanced Purification Technology for Ceftizoxime Sodium: Scaling High-Purity Antibiotic Production
The pharmaceutical industry continuously demands higher purity standards for critical antibiotics to ensure patient safety and therapeutic efficacy. Patent CN102079750B introduces a groundbreaking purification method for Ceftizoxime Sodium, a third-generation cephalosporin known commercially as SKF-88373. This technology addresses the persistent challenges of impurity profiles and stability that have long plagued the manufacturing of this essential respiratory and urinary tract infection treatment. By integrating a specific acid-base reaction sequence with optimized activated carbon adsorption and preparative high-performance liquid chromatography (HPLC), the process achieves a substantial leap in quality control. The method ensures that the final bulk drug meets stringent purity specifications, significantly reducing the risk of toxic side effects associated with lower-grade intermediates. For R&D directors and procurement specialists, understanding this patented approach is vital for securing a reliable antibiotic supplier capable of delivering consistent, high-quality active pharmaceutical ingredients. The innovation lies not just in the chemical transformation but in the meticulous separation physics that allow for the isolation of the target molecule from complex reaction byproducts.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for Ceftizoxime Sodium often rely heavily on simple crystallization or less efficient chromatographic media, which fail to adequately remove closely related impurities and colored substances. Prior art methods, such as those utilizing alumina columns or macroporous resins, frequently suffer from issues regarding rigidity, fragmentation, and the potential for secondary pollution due to residual pore-creating agents. These conventional techniques often result in products with lower purity, poor clarity upon dissolution, and compromised stability in aqueous solutions, which are critical failures for injectable formulations. Furthermore, older synthetic routes involving multiple deprotection steps or direct condensation without rigorous purification tend to generate significant amounts of side reactions, necessitating complex and costly downstream processing. The inability to consistently achieve purity levels above 99% using these legacy methods creates a bottleneck for manufacturers aiming to meet modern regulatory standards for parenteral antibiotics. Consequently, the yield is often sacrificed in an attempt to improve purity, leading to inefficient resource utilization and higher overall production costs for the supply chain.
The Novel Approach
The novel approach disclosed in the patent fundamentally reengineers the purification workflow by optimizing the stationary phase and mobile phase parameters to maximize separation efficiency. Instead of relying on standard alumina or resin columns, this method utilizes specific silica gel with controlled particle diameters ranging from 45-250 μm and pore sizes of 20-30 Å, which provides a superior surface area for interaction. The process incorporates a precise activated carbon treatment step, where the dosage is strictly controlled between 0.1% and 0.3% (g/ml) to remove color bodies without irreversibly adsorbing the active pharmaceutical ingredient. By employing a water and acetonitrile mixed solvent system in a 1:3 volume ratio as the mobile phase, the method ensures a smooth elution profile that effectively separates the target compound from impurities. This refined technique allows for the collection of fractions with a content greater than 90%, which are then merged to produce a final product with purity exceeding 99.5%. The result is a robust, scalable process that offers high yield and exceptional quality, making it highly suitable for industrial production and meeting the needs of a reliable agrochemical intermediate supplier or pharma partner.
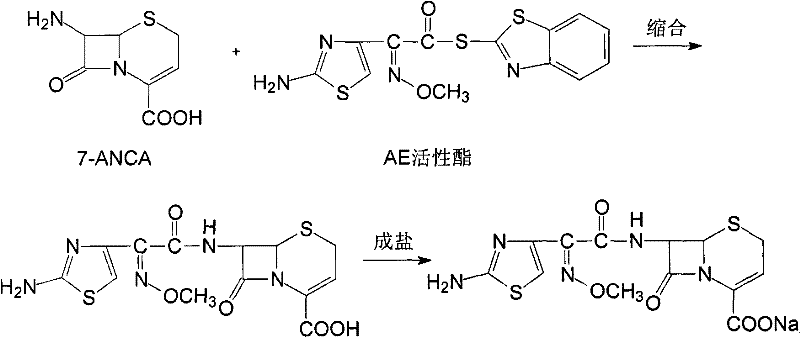
Mechanistic Insights into Silica Gel Chromatographic Purification
The core of this technological advancement lies in the physicochemical interactions between the Ceftizoxime molecule and the optimized silica gel stationary phase. Silica gel, with its specific surface area and pore distribution, facilitates a more uniform adsorption-desorption cycle compared to the irregular structures of alumina or macroporous resins. The mechanism involves the differential partitioning of the Ceftizoxime acid and its impurities between the polar silica surface and the organic-aqueous mobile phase. By maintaining the column temperature between 30-40°C and controlling the flow velocity at 2.8-4.5 ml/min, the system minimizes band broadening and ensures sharp separation peaks. This precise control over chromatographic conditions prevents the co-elution of structurally similar byproducts, which is a common failure mode in less optimized systems. The use of a specific mobile phase composition further enhances the selectivity, allowing the target molecule to migrate at a distinct rate while retaining impurities longer on the column. This mechanistic precision is what enables the consistent production of high-purity Ceftizoxime Sodium, a critical factor for any high-purity OLED material or pharmaceutical intermediate manufacturer seeking quality assurance.
Impurity control is further enhanced through the strategic application of activated carbon adsorption prior to chromatography. The mechanism here involves the physical adsorption of large molecular weight colored impurities and organic particulates onto the high-surface-area carbon pores. By strictly limiting the carbon dosage to 0.1%-0.3%, the process avoids the common pitfall of product loss, where excessive carbon adsorbs the valuable active ingredient along with the impurities. Following this, the acid-base reaction steps serve to convert the compound between its acid and salt forms, exploiting solubility differences to precipitate out remaining inorganic salts and soluble impurities. The final alkalization step, adjusting the pH to between 8 and 13 using sodium bicarbonate or sodium hydroxide, ensures the complete conversion to the sodium salt form while precipitating the product in a highly crystalline state. This multi-barrier approach to impurity removal ensures that the final impurity profile is drastically simplified, meeting the rigorous demands of regulatory bodies for injectable antibiotics.
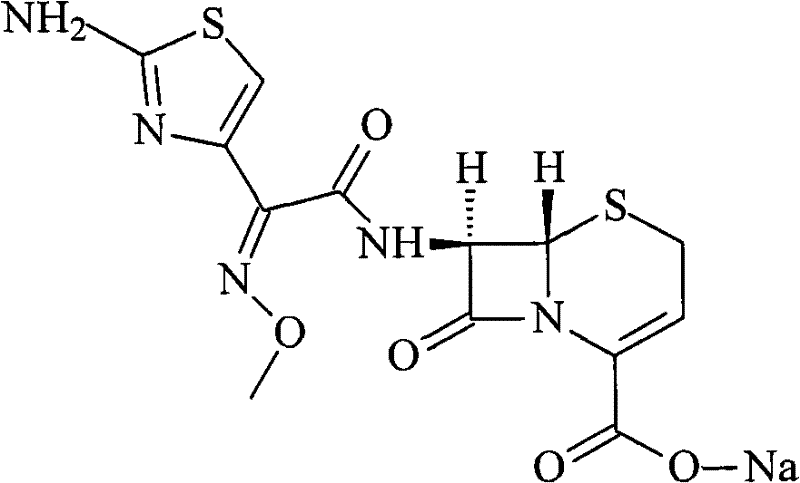
How to Synthesize Ceftizoxime Sodium Efficiently
The synthesis and purification of Ceftizoxime Sodium require a disciplined approach to process parameters to ensure reproducibility and high yield on a commercial scale. The patented method outlines a clear sequence starting from the bulk drug crude, moving through acidification, decolorization, chromatographic separation, and final salt formation. Each step is critical, from the selection of the organic acid for precipitation to the specific ratio of the water-acetonitrile mobile phase. Operators must adhere strictly to the temperature and flow rate specifications to maintain the integrity of the beta-lactam ring and prevent degradation. The following guide summarizes the standardized synthesis steps derived from the patent data, providing a roadmap for technical teams to implement this high-efficiency route. Detailed standardized synthesis steps are provided in the guide below for immediate operational reference.
- Dissolve crude Ceftizoxime Sodium in purified water and adjust pH to 2-4 using organic acid to precipitate Ceftizoxime acid.
- Dissolve the acid in organic solvent, add 0.1%-0.3% activated carbon, stir at 40-50°C, and filter to remove impurities.
- Purify the filtrate using a silica gel chromatographic column with a water-acetonitrile mobile phase, then adjust pH to 8-13 to precipitate the final high-purity salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this purification technology translates into tangible strategic advantages regarding cost stability and supply continuity. The elimination of complex deprotection steps and the use of readily available silica gel instead of specialized resins significantly reduces the raw material costs associated with the purification media. Furthermore, the high yield achieved through optimized carbon usage and precise fraction collection means that less starting material is wasted, directly improving the overall material balance of the production line. The robustness of the process reduces the likelihood of batch failures, which is a major cost driver in pharmaceutical manufacturing, thereby ensuring a more predictable supply of the active ingredient. By simplifying the workflow and enhancing the purity in a single pass, the need for reprocessing or secondary purification is drastically reduced, leading to substantial cost savings in labor and energy consumption. This efficiency makes the technology highly attractive for cost reduction in electronic chemical manufacturing or pharmaceutical contexts where margin pressure is high.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive and complex chromatographic media like macroporous resins, replacing them with cost-effective silica gel that offers superior performance and longevity. By optimizing the activated carbon dosage, the loss of valuable product during the decolorization phase is minimized, which directly improves the overall yield and reduces the cost per kilogram of the final API. The simplified workflow reduces the number of unit operations required, leading to lower energy consumption and reduced labor hours per batch. Additionally, the high purity achieved reduces the regulatory risk and potential costs associated with batch rejection or recall due to impurity failures. These factors combine to create a significantly more economical production model compared to legacy methods.
- Enhanced Supply Chain Reliability: The use of common and readily available reagents such as silica gel, acetonitrile, and standard organic acids ensures that the supply chain is not vulnerable to shortages of specialized catalysts or proprietary resins. The robustness of the process parameters, such as the wide acceptable range for column temperature and flow rate, allows for greater flexibility in manufacturing scheduling and equipment utilization. This flexibility reduces the lead time for high-purity pharmaceutical intermediates, enabling faster response to market demand fluctuations. The high stability of the final product also implies a longer shelf life and reduced waste during storage and transportation, further securing the supply chain against logistical disruptions. Partners can rely on a consistent output quality that minimizes the need for incoming quality control interventions.
- Scalability and Environmental Compliance: The method is designed with industrial scale-up in mind, utilizing standard chromatographic equipment and solvent systems that are easily managed in large-scale production facilities. The reduction in side reactions and the efficient separation of impurities mean that the waste stream is cleaner and easier to treat, aligning with strict environmental compliance standards. The process avoids the use of heavy metal catalysts or hazardous reagents that would require complex waste disposal procedures, thereby reducing the environmental footprint of the manufacturing site. The ability to scale from laboratory to commercial production without significant process re-engineering ensures a smooth transition for commercial scale-up of complex polymer additives or pharmaceutical compounds. This scalability ensures that supply can be ramped up quickly to meet global demand without compromising on quality or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this purification technology. These answers are derived directly from the patent specifications and experimental data to provide accurate guidance for technical teams. Understanding these details is crucial for evaluating the feasibility of integrating this process into existing manufacturing lines. The insights provided here help clarify the operational benefits and technical requirements for successful adoption.
Q: What is the primary advantage of the chromatographic method in CN102079750B?
A: The primary advantage is the significant improvement in product purity, achieving levels greater than 99.5%, while maintaining a high yield compared to traditional crystallization methods which often struggle with impurity removal.
Q: Why is silica gel preferred over alumina for this purification process?
A: Silica gel is preferred because it offers superior controlled physical structure regarding particle size and pore distribution, leading to better separation efficiency and higher finished product purity compared to alumina or macroporous resin.
Q: How does this process impact the stability of the final antibiotic product?
A: By effectively removing colored impurities and related substances through activated carbon adsorption and precise chromatographic separation, the process ensures the final Ceftizoxime Sodium has enhanced stability in aqueous solutions and reduced toxic side effects.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ceftizoxime Sodium Supplier
NINGBO INNO PHARMCHEM stands at the forefront of chemical manufacturing, leveraging advanced purification technologies like the one described in CN102079750B to deliver superior pharmaceutical intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Ceftizoxime Sodium meets the highest global standards for safety and efficacy. Our commitment to technical excellence allows us to navigate complex synthesis routes, providing our partners with a reliable source of high-quality active ingredients that drive their own product success. By choosing us, you are partnering with a team that understands the critical importance of purity and stability in the healthcare sector.
We invite you to engage with our technical procurement team to discuss how we can optimize your supply chain for Ceftizoxime Sodium and other critical intermediates. Request a Customized Cost-Saving Analysis to understand how our efficient manufacturing processes can reduce your overall procurement costs without compromising quality. Our experts are ready to provide specific COA data and route feasibility assessments to support your R&D and production planning. Let us help you secure a stable, high-quality supply of essential antibiotics for your global operations.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →