Advanced Synthesis of LE14 Linker Drug Conjugates for Commercial ADC Manufacturing
The pharmaceutical industry is witnessing a paradigm shift in the development of Antibody Drug Conjugates (ADCs), with Linker Drug conjugates like LE14 serving as critical payloads for targeted cancer therapies. Patent CN116178386A discloses a groundbreaking preparation method that addresses long-standing stability and purity challenges in the synthesis of these complex molecules. By introducing a novel carboxylic acid intermediate strategy, this technology enables the production of high-purity LE14 while significantly streamlining the manufacturing workflow. For R&D Directors and Supply Chain Heads, this represents a viable pathway to overcome the purification bottlenecks that have historically plagued ADC intermediate production. The method leverages the economic advantages of Exatecan mesylate over expensive DXd derivatives, ensuring a more robust and cost-effective supply chain for next-generation oncology treatments.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
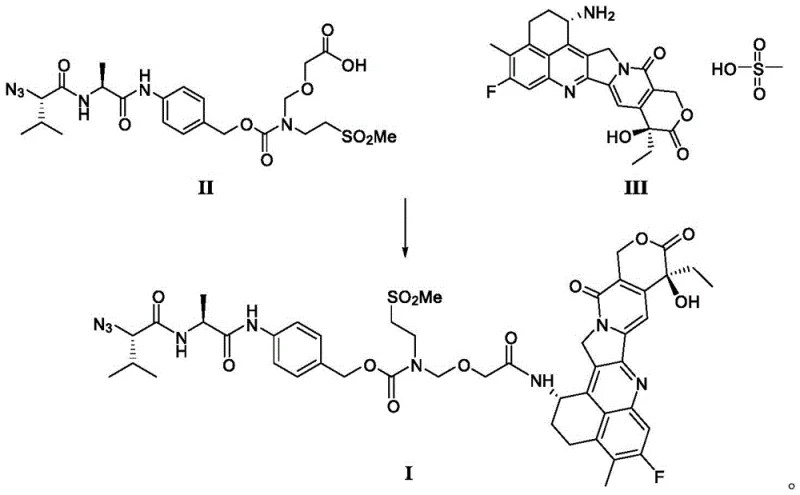
Historically, the synthesis of LE14 and similar linker drug conjugates has been hindered by the instability of key carboxylic acid intermediates. In conventional Routes 1 and 2, intermediates such as 1-4 and 2-2a are generated via tert-butyl deprotection using trifluoroacetic acid. These intermediates are notoriously unstable under acidic and alkaline conditions, leading to significant decomposition during purification processes. Consequently, manufacturers are forced to use crude products directly in subsequent steps, which introduces persistent impurities that are difficult to remove due to their similar polarity to the final product. Furthermore, Routes 3, 4, and 5 rely on expensive DXd derivatives as payload sources, which drastically inflates the raw material costs and complicates the supply chain logistics for large-scale commercial production.
The Novel Approach
The innovative method described in CN116178386A circumvents these issues by utilizing a stable Intermediate II as the starting material for process production. This intermediate is protected with a trimethylsilylethyl group, which withstands the rigors of purification and allows for the isolation of a high-purity carboxylic acid product. By stabilizing this critical junction in the synthesis, the process significantly reduces the impurity content in the final LE14 product. Additionally, the route employs the more economical Exatecan mesylate for the condensation step, bypassing the need for costly pre-derivatized DXd payloads. This strategic shift not only simplifies the process steps but also enhances the overall feasibility of industrial-scale manufacturing for reliable ADC linker supplier operations.
Mechanistic Insights into Fluoride-Mediated Deprotection and Amide Condensation
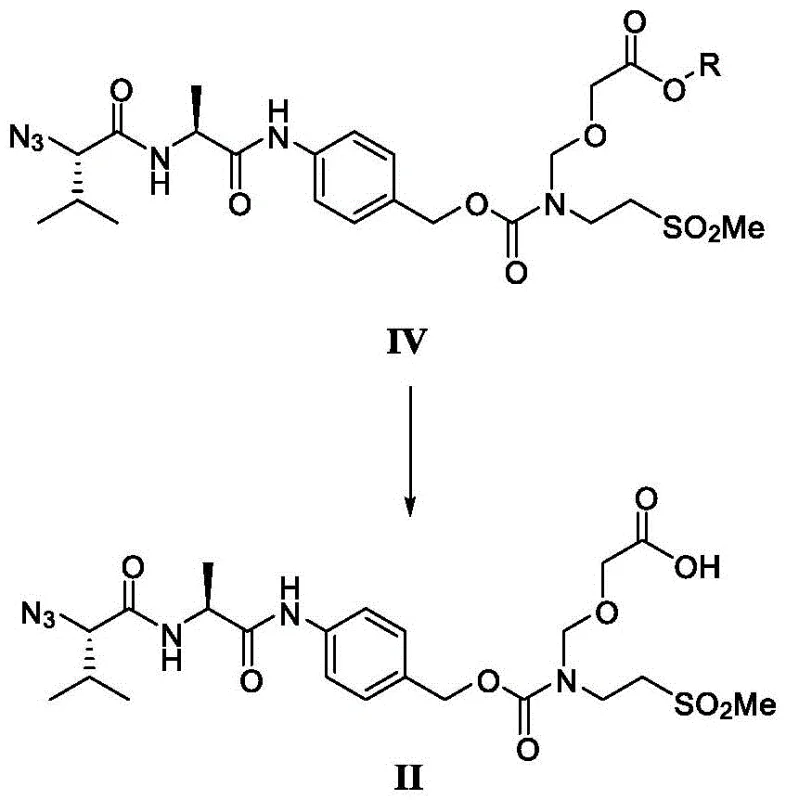
The core of this technological breakthrough lies in the precise control of the deprotection and condensation mechanisms. The synthesis of Intermediate II involves a fluoride-mediated deprotection of the silyl-protected precursor (Compound IV). Unlike traditional acid-labile protecting groups, the trimethylsilylethyl moiety is selectively removed using fluoride reagents such as tetrabutylammonium fluoride or potassium fluoride under mild conditions. This specificity prevents the degradation of the sensitive peptide backbone and the azide functionality, which are prone to side reactions under harsh acidic conditions. The resulting carboxylic acid Intermediate II is chemically stable, allowing for rigorous quality control via silica gel column chromatography before it enters the condensation phase.
Following the isolation of Intermediate II, the amide condensation with Exatecan mesylate (Compound III) is executed using advanced coupling agents like DMTMM. This reaction is conducted under inert gas protection at controlled temperatures to minimize racemization and side-product formation. The use of a purified Intermediate II ensures that the condensation proceeds with high efficiency, drastically reducing the generation of difficult-to-remove impurities. This mechanistic precision is crucial for achieving the stringent purity specifications required for clinical-grade ADC intermediates, ensuring that the final LE14 product meets the rigorous standards demanded by global regulatory bodies for high-purity ADC intermediates.
How to Synthesize LE14 Efficiently
The synthesis of LE14 via this novel route involves a sequence of highly controlled chemical transformations designed to maximize yield and purity. The process begins with the substitution of an azide polypeptide compound to introduce the chloromethyl functionality, followed by etherification with a silyl-protected glycolate reagent. The critical step involves the fluoride-mediated deprotection to generate the stable carboxylic acid Intermediate II, which is then purified to remove any trace impurities. This purified intermediate is subsequently condensed with Exatecan mesylate, reduced to the amine, and finally coupled with the maleimide linker to yield the final LE14 conjugate. Detailed standardized synthesis steps are provided in the guide below to ensure reproducibility and compliance with GMP standards.
- Perform substitution reaction on azide polypeptide compound VII with paraformaldehyde and trimethylchlorosilane to obtain compound VI.
- Conduct etherification reaction on compound VI with silyl-protected glycolate reagent V to obtain stable intermediate IV.
- Deprotect intermediate IV using a fluoride reagent to obtain purified carboxylic acid intermediate II, then condense with Exatecan mesylate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers substantial strategic benefits beyond mere technical performance. The primary advantage lies in the significant cost reduction in ADC manufacturing achieved by substituting expensive DXd derivatives with economically viable Exatecan mesylate. This raw material optimization directly impacts the bill of materials, allowing for more competitive pricing structures without compromising on quality. Furthermore, the ability to purify Intermediate II means that quality issues are addressed early in the process, reducing the risk of batch failures and ensuring a more reliable ADC linker supplier partnership for long-term projects.
- Cost Reduction in Manufacturing: The elimination of expensive DXd derivatives from the early stages of synthesis results in substantial cost savings. By utilizing Exatecan mesylate, which is more readily available and economical, the overall production cost is drastically simplified. This approach avoids the complex and loss-prone derivatization steps associated with irinotecan-to-DXd conversion seen in prior art, thereby optimizing the utilization of high-value starting materials and enhancing the economic viability of commercial scale-up of complex linker drug conjugates.
- Enhanced Supply Chain Reliability: The stability of Intermediate II allows for stockpiling and quality verification before proceeding to the final coupling steps. This decoupling of the synthesis stages reduces the pressure on just-in-time manufacturing and mitigates the risk of supply disruptions caused by unstable intermediates. By ensuring that key building blocks are stable and purifiable, the lead time for high-purity ADC intermediates is effectively managed, providing a more predictable and secure supply chain for downstream antibody conjugation processes.
- Scalability and Environmental Compliance: The use of milder deprotection conditions and the ability to purify intermediates via standard column chromatography facilitates easier scale-up from laboratory to commercial production. The process avoids the use of harsh acids like trifluoroacetic acid in critical purification steps, which simplifies waste treatment and aligns with stricter environmental compliance standards. This scalability ensures that the manufacturing process can be expanded to meet growing market demand for ADC therapies without encountering the technical bottlenecks typical of older synthesis routes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel LE14 synthesis route. These insights are derived directly from the patent data and are intended to clarify the advantages of this method over conventional approaches. Understanding these details is essential for technical teams evaluating the feasibility of adopting this process for their own manufacturing pipelines or for procurement teams assessing the long-term value of this technology.
Q: Why is Intermediate II more stable than previous carboxylic acid intermediates?
A: Unlike intermediates 1-4 and 2-2a in prior routes which decompose during purification, Intermediate II utilizes a trimethylsilylethyl protecting group that allows for stable isolation and column chromatography purification, significantly reducing impurities.
Q: How does this route reduce production costs for ADC payloads?
A: The process utilizes economically available Exatecan mesylate directly for condensation, avoiding the need for expensive pre-derivatized DXd payloads required in conventional Routes 3, 4, and 5, thereby lowering raw material costs.
Q: What purity levels can be achieved with this novel synthesis method?
A: The method enables the production of final LE14 product with purity exceeding 99.5% and maximum single impurity content below 0.1%, surpassing the purity profiles of prior art routes which often struggle with impurity removal.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable LE14 Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthesis routes in the development of next-generation ADC therapies. Our CDMO expertise allows us to implement complex pathways like the one described in CN116178386A with precision and reliability. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from clinical trials to market launch. Our rigorous QC labs and stringent purity specifications guarantee that every batch of LE14 meets the highest international standards, providing you with a secure foundation for your drug development programs.
We invite you to collaborate with us to optimize your supply chain and reduce manufacturing costs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific project needs. By partnering with us, you gain access to specific COA data and route feasibility assessments that can accelerate your development timeline. Contact us today to discuss how our advanced synthesis capabilities can support your goal of delivering high-quality ADC therapies to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →