Advanced Synthesis of ADC Linker-Drug Conjugates: Technical Upgrades and Commercial Scalability
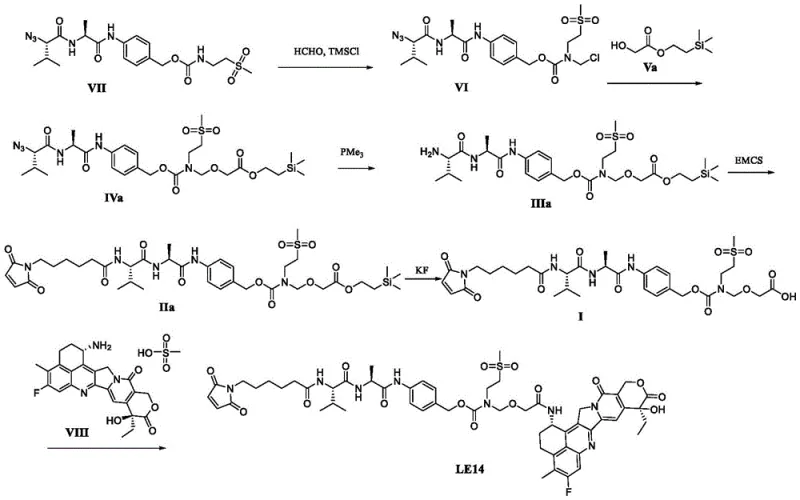
The rapidly evolving landscape of Antibody-Drug Conjugates (ADCs) demands unprecedented precision in the synthesis of linker-drug components, where stability and purity are paramount for clinical success. Patent CN116217655A introduces a transformative preparation method for a key intermediate, designated as Formula I, which serves as the critical bridge in the construction of Compound LE, specifically the potent LE14 conjugate. This technological breakthrough addresses the longstanding industry challenge of intermediate instability during purification, a bottleneck that has historically plagued the scalable manufacturing of complex ADC payloads. By establishing a route where the carboxylic acid intermediate remains chemically robust throughout downstream processing, this innovation not only enhances the structural integrity of the final molecule but also streamlines the operational workflow for reliable ADC intermediate supplier networks globally. The strategic implementation of this chemistry allows for the production of high-purity materials essential for next-generation oncology therapeutics.
In the realm of modern oncology drug development, the ability to consistently produce linker-drug conjugates with minimal impurity profiles is a decisive factor for regulatory approval and patient safety. The methodology outlined in this patent leverages a sophisticated deprotection strategy followed by high-pressure preparative chromatography, ensuring that the resulting Formula I compound exceeds 95.0% purity before it ever encounters the cytotoxic warhead. This level of quality control is indispensable for cost reduction in linker-drug manufacturing, as it minimizes the loss of expensive starting materials like Exatecan in later stages. For procurement and technical teams, understanding the nuances of this synthesis provides a competitive edge in securing supply chains that are both resilient and compliant with rigorous pharmaceutical standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical approaches to synthesizing ADC linker intermediates, such as those depicted in Routes 1 and 2 of the prior art, suffer from inherent chemical instabilities that compromise both yield and product quality. In these conventional pathways, the carboxylic acid intermediates (such as 1-4 and 2-2a) exhibit significant sensitivity to acidic and basic conditions, leading to unavoidable decomposition during the critical purification phases. This instability forces manufacturers to either accept lower purity grades or utilize the crude material directly in subsequent steps, which inevitably propagates impurities into the final drug substance. Such impurities often possess polarities strikingly similar to the target molecule, rendering them exceptionally difficult to remove via standard chromatographic techniques and ultimately jeopardizing the safety profile of the ADC. Furthermore, the inability to effectively purify these intermediates results in substantial material loss, driving up the cost of goods and creating supply chain vulnerabilities for high-purity pharmaceutical intermediates.
The Novel Approach
In stark contrast, the novel approach presented in CN116217655A utilizes a specially designed Formula I intermediate that demonstrates exceptional stability throughout the purification process, effectively eliminating the decomposition risks associated with earlier methodologies. This robustness allows for the application of high-pressure preparative chromatography, a technique capable of resolving complex impurity profiles that conventional low-pressure columns cannot address. By securing a highly pure intermediate before the introduction of the cytotoxic payload, the process ensures that the final coupling reaction proceeds with maximum efficiency and minimal side reactions. This strategic shift not only improves the overall yield of the final LE14 conjugate but also simplifies the downstream processing requirements, making the entire manufacturing sequence more amenable to commercial scale-up of complex ADC intermediates. The result is a streamlined production capability that aligns perfectly with the demanding timelines of modern drug development pipelines.
Mechanistic Insights into Fluoride-Mediated Deprotection and Amide Coupling
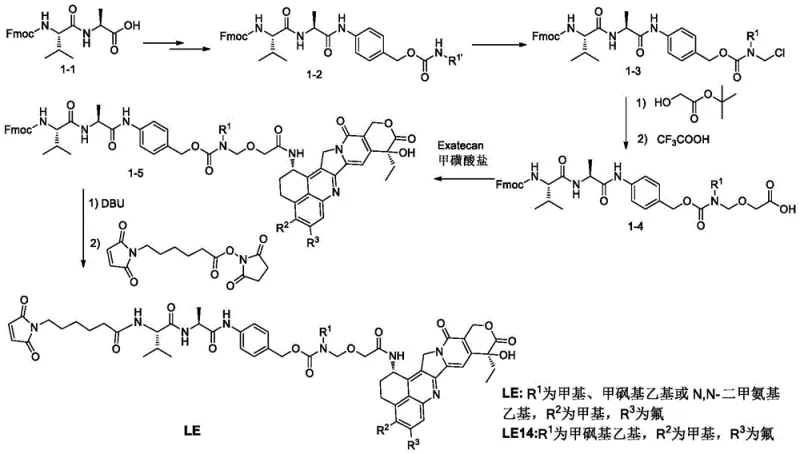
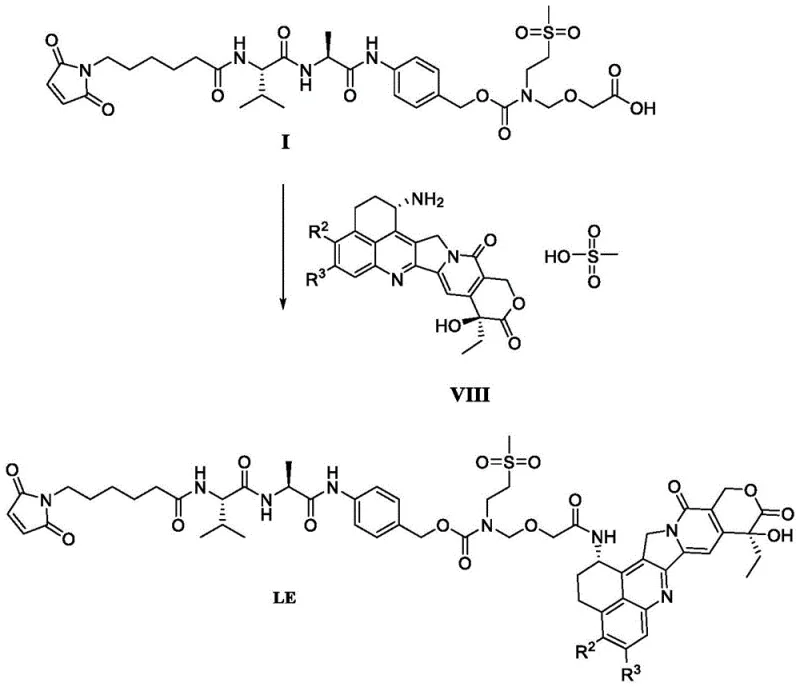
The core chemical innovation lies in the deprotection of the silyl-protected precursor (Formula II) to generate the free carboxylic acid (Formula I) using specific fluorine reagents under controlled thermal conditions. This transformation is meticulously optimized to proceed at temperatures between 50°C and 70°C, utilizing reagents such as potassium fluoride or tetrabutylammonium fluoride in polar aprotic solvents like DMF. The mechanistic precision here ensures that the cleavage of the silyl ether bond occurs cleanly without affecting other sensitive functional groups within the complex linker structure, such as the maleimide moiety or the peptide bonds. Following this deprotection, the crude intermediate undergoes a rigorous purification regimen involving dynamic axial compression columns packed with unbonded silanol stationary phases. This high-resolution separation technique is critical for removing trace impurities that could otherwise interfere with the subsequent conjugation chemistry, thereby safeguarding the integrity of the final therapeutic agent.
Following the isolation of high-purity Formula I, the synthesis culminates in an amide condensation reaction with Exatecan mesylate (Formula VIII), a step that requires precise stoichiometric control and the use of advanced coupling agents like DMTMM. This coupling is performed under mild conditions, typically between 20°C and 30°C, to preserve the stereochemical integrity of the chiral centers within the linker and the payload. The choice of DMTMM as a condensing agent is particularly advantageous as it minimizes racemization and suppresses the formation of urea byproducts common with carbodiimide reagents. By delaying the introduction of the cytotoxic Exatecan until this final stage, the process significantly reduces lead time for high-purity pharmaceutical intermediates related to safety handling, as the bulk of the synthesis involves non-toxic materials. This late-stage functionalization strategy is a hallmark of efficient ADC manufacturing, balancing chemical complexity with operational safety and economic viability.
How to Synthesize LE14 Efficiently
The synthesis of Compound LE14 begins with the preparation of the stable Formula I intermediate, followed by its activation and coupling with the cytotoxic payload. Detailed standardized synthesis steps see the guide below. This process is designed to maximize yield while maintaining the stringent purity specifications required for clinical applications. The operational parameters, including solvent ratios, reaction times, and temperature controls, are optimized to ensure reproducibility across different batch sizes, from laboratory scale to multi-kilogram production runs.
- Deprotect Compound of Formula II using a fluorine reagent such as potassium fluoride in DMF at 60°C to obtain crude Formula I.
- Purify the crude Formula I using high-pressure preparative chromatography with a UniSil stationary phase to achieve purity greater than 95.0%.
- Perform amide condensation between purified Formula I and Exatecan mesylate using DMTMM as a condensing agent to yield Compound LE14.
Commercial Advantages for Procurement and Supply Chain Teams
The adoption of this advanced synthesis route offers profound commercial advantages that extend beyond mere chemical elegance, directly impacting the bottom line and operational reliability of pharmaceutical manufacturing. By stabilizing the key intermediate, the process eliminates the need for costly rework or disposal of degraded batches, leading to substantial cost savings in raw material utilization. The ability to purify the intermediate to over 95% purity before payload conjugation means that the expensive cytotoxic drug substance is not wasted on flawed linker molecules, thereby optimizing the utilization rate of high-value inputs. For supply chain leaders, this translates into a more predictable production schedule with fewer interruptions caused by quality failures, ensuring a continuous flow of critical materials for downstream antibody conjugation processes.
- Cost Reduction in Manufacturing: The enhanced stability of Formula I allows for the use of high-pressure chromatography which, while capital intensive, delivers significantly higher recovery rates compared to the low-yield purification of unstable intermediates in prior art. This improvement in material throughput directly lowers the cost per gram of the final API intermediate. Furthermore, by avoiding the use of pre-derivatized, expensive DXd payloads found in other routes and instead coupling Exatecan at the end, the process leverages a more cost-effective starting material strategy. The elimination of decomposition losses during purification means that less starting material is required to produce the same amount of final product, driving down the overall variable costs associated with large-scale production.
- Enhanced Supply Chain Reliability: A stable intermediate that does not degrade during storage or processing provides a crucial buffer against supply chain disruptions. Manufacturers can stockpile purified Formula I with confidence, knowing it will retain its specification over time, which facilitates better inventory management and just-in-time delivery models. The robustness of the chemistry also reduces the dependency on highly specialized, low-throughput purification methods that often become bottlenecks in commercial manufacturing. This reliability ensures that downstream conjugation facilities receive consistent, high-quality inputs, minimizing the risk of batch failures in the final drug product and securing the continuity of supply for clinical trials and commercial launches.
- Scalability and Environmental Compliance: The process is designed with industrial scalability in mind, utilizing reagents and solvents that are manageable in large reactor systems without requiring exotic conditions. The high efficiency of the purification step reduces the volume of solvent waste generated per unit of product, aligning with green chemistry principles and reducing the environmental footprint of the manufacturing site. Additionally, the late-stage introduction of the cytotoxin minimizes the containment requirements for the majority of the synthesis steps, lowering the operational complexity and cost of facility maintenance. This combination of scalability and environmental stewardship makes the technology highly attractive for long-term commercial partnerships and regulatory compliance.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of this ADC linker technology. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on performance metrics and operational benefits. Understanding these details is crucial for technical teams evaluating this route for potential integration into their manufacturing portfolios.
Q: Why is the new Formula I intermediate considered more stable than previous carboxylic acid intermediates?
A: Unlike intermediates in Routes 1 and 2 which decompose during purification due to acidity and alkalinity instability, Formula I remains stable throughout the purification process, allowing for high-pressure chromatography without degradation.
Q: How does this synthesis route improve safety regarding cytotoxic substances?
A: The process delays the introduction of the cytotoxic payload (Exatecan) until the final coupling step. This significantly reduces the exposure time of personnel to toxic materials during the synthesis of the linker intermediate.
Q: What purity levels can be achieved for the final LE14 product using this method?
A: By utilizing high-pressure preparative chromatography for the intermediate, the final Compound LE14 can achieve a purity of 99.17% with a single impurity content as low as 0.21%, meeting stringent IND declaration requirements.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable LE14 Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of ADC therapies hinges on the availability of high-quality linker-drug intermediates produced via robust and scalable processes. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from clinical phases to market launch. Our facilities are equipped with state-of-the-art high-pressure purification systems and rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of LE14 or custom linker intermediate adheres to the highest global standards. We are committed to delivering not just chemicals, but reliable solutions that de-risk your supply chain.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this stable intermediate platform. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing you to validate the superior purity and yield profiles demonstrated in patent CN116217655A. Let us collaborate to accelerate your ADC development timeline with a supply partner dedicated to excellence and innovation.