Advanced Manufacturing of Linker Drug Conjugate LE14 via Stable Intermediate Strategy
Introduction to Advanced ADC Linker Technology
The rapidly evolving landscape of Antibody-Drug Conjugates (ADCs) demands increasingly sophisticated linker technologies that balance stability in circulation with efficient release at the tumor site. Patent CN116217654A introduces a groundbreaking preparation method for a linker drug conjugate, specifically targeting the synthesis of LE14 and its critical intermediates. This innovation addresses long-standing challenges in the field, particularly regarding the stability of carboxylic acid intermediates and the economic burden of early-stage payload attachment. By re-engineering the synthetic pathway, this technology enables the production of high-purity linker drug conjugates suitable for clinical applications. The methodology focuses on generating stable intermediates, such as Formula I, which can be purified effectively before the final conjugation step. This strategic shift not only enhances the quality profile of the final Active Pharmaceutical Ingredient (API) but also streamlines the manufacturing process, making it a viable option for reliable linker drug conjugate supplier networks aiming for industrial scalability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex ADC linkers like LE14 has been plagued by significant technical hurdles that impact both yield and cost efficiency. As illustrated in prior art routes such as Route 3, traditional methods often rely on the early introduction of expensive cytotoxic payloads like DXd or its derivatives. 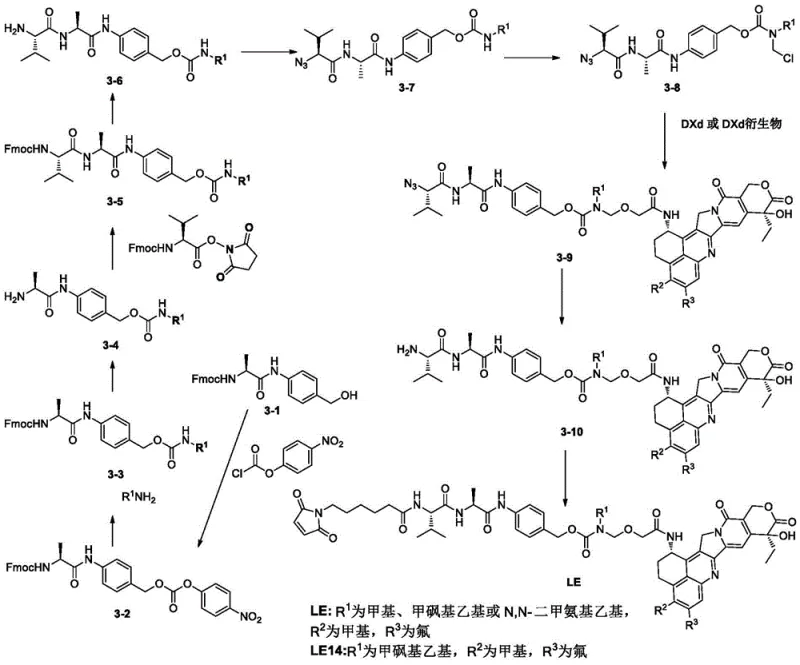 This approach creates a bottleneck where valuable, high-cost materials are subjected to multiple subsequent reaction steps, leading to substantial cumulative losses. Furthermore, conventional pathways frequently generate carboxylic acid intermediates that are chemically unstable under standard purification conditions. For instance, in Route 1 and Route 2, intermediates like 1-4 and 2-2a decompose during deprotection and purification, forcing manufacturers to use crude materials for downstream reactions. This lack of purification capability results in the carryover of impurities with polarities similar to the target product, drastically complicating final isolation and failing to meet stringent regulatory purity standards required for IND filings. These inefficiencies collectively drive up the cost reduction in ADC manufacturing barriers and limit supply chain reliability.
This approach creates a bottleneck where valuable, high-cost materials are subjected to multiple subsequent reaction steps, leading to substantial cumulative losses. Furthermore, conventional pathways frequently generate carboxylic acid intermediates that are chemically unstable under standard purification conditions. For instance, in Route 1 and Route 2, intermediates like 1-4 and 2-2a decompose during deprotection and purification, forcing manufacturers to use crude materials for downstream reactions. This lack of purification capability results in the carryover of impurities with polarities similar to the target product, drastically complicating final isolation and failing to meet stringent regulatory purity standards required for IND filings. These inefficiencies collectively drive up the cost reduction in ADC manufacturing barriers and limit supply chain reliability.
The Novel Approach
In stark contrast, the novel approach disclosed in CN116217654A fundamentally restructures the synthetic logic to prioritize intermediate stability and late-stage functionalization. The core innovation lies in the design of a robust carboxylic acid intermediate, designated as Formula I, which remains stable throughout the purification process. This stability allows for rigorous chromatographic purification, ensuring that impurities are removed before the costly payload is introduced. 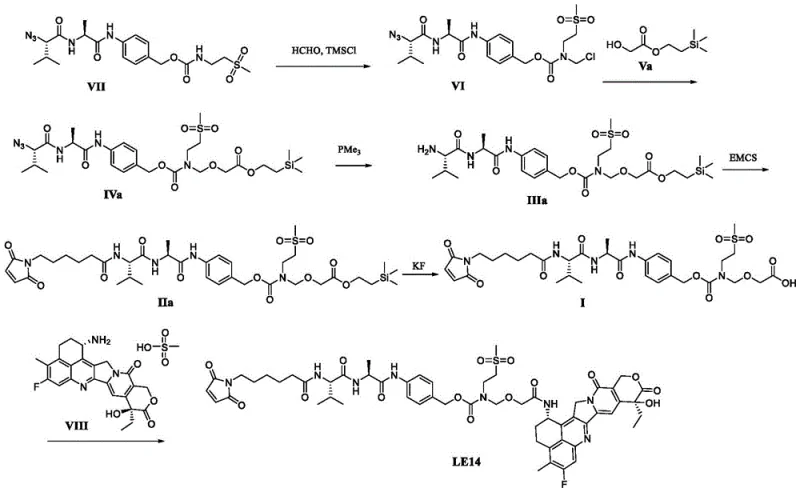 By deferring the conjugation of the cytotoxic payload (Exatecan) to the final step, the process minimizes the exposure of operators to hazardous materials and prevents the degradation of expensive drug components during multi-step synthesis. The route utilizes a sequence involving azide reduction and maleimide coupling that is highly controlled and scalable. This methodological shift transforms the production of high-purity linker drug conjugates from a fragile, low-yield operation into a robust, commercially viable process. It effectively decouples the complex linker synthesis from the payload handling, offering a modular approach that enhances both safety and economic feasibility for large-scale production facilities.
By deferring the conjugation of the cytotoxic payload (Exatecan) to the final step, the process minimizes the exposure of operators to hazardous materials and prevents the degradation of expensive drug components during multi-step synthesis. The route utilizes a sequence involving azide reduction and maleimide coupling that is highly controlled and scalable. This methodological shift transforms the production of high-purity linker drug conjugates from a fragile, low-yield operation into a robust, commercially viable process. It effectively decouples the complex linker synthesis from the payload handling, offering a modular approach that enhances both safety and economic feasibility for large-scale production facilities.
Mechanistic Insights into Azide Reduction and Maleimide Coupling
The chemical elegance of this synthesis is anchored in two critical transformations: the selective reduction of the azide group and the subsequent bioconjugation via maleimide chemistry. The conversion of the azide-containing precursor (Formula IV) to the amine intermediate (Formula III) is achieved using a phosphine-based reducing agent, such as trimethylphosphine, in the presence of an acid buffer. This Staudinger-like reduction is meticulously optimized to proceed at mild temperatures, typically between 0°C and 20°C, preventing side reactions that could compromise the integrity of the sensitive peptide backbone. The use of an acid buffer, specifically sodium acetate at pH 5.0, is crucial for protonating the intermediate iminophosphorane, facilitating hydrolysis to the free amine without inducing racemization or degradation of adjacent chiral centers. Following the generation of the free amine, the process employs 6-(maleimido)hexanoic acid succinimidyl ester (EMCS) for coupling. This reaction forms a stable amide bond while preserving the maleimide moiety for future antibody conjugation. The reaction conditions are carefully tuned, utilizing solvents like dichloromethane and maintaining temperatures around 30°C-35°C to maximize conversion while minimizing hydrolysis of the activated ester. This precise control over reaction kinetics ensures that the resulting Formula II compound possesses the structural fidelity necessary for downstream therapeutic efficacy.
Impurity control is another pillar of this mechanistic strategy, particularly concerning the final deprotection and conjugation steps. The removal of the silyl protecting group from Formula II to yield the key carboxylic acid Formula I is performed using fluoride sources like potassium fluoride in DMF. Unlike previous methods where acidic deprotection led to decomposition, this fluoride-mediated cleavage is orthogonal and mild, preserving the acid-labile groups within the molecule. The resulting carboxylic acid is then activated using coupling reagents like DMTMM for the final attachment of Exatecan mesylate. This activation method is superior to traditional carbodiimides as it generates fewer urea byproducts that are difficult to remove. The entire sequence is designed to minimize the formation of diastereomers and deletion sequences, which are common pitfalls in peptide-based linker synthesis. By maintaining a closed system under inert gas protection and utilizing high-purity reagents, the process ensures that the final LE14 product meets the rigorous specification of having total impurities below 3% and single impurities below 1%, a benchmark essential for clinical grade materials.
How to Synthesize LE14 Efficiently
The synthesis of LE14 described in this patent represents a paradigm shift towards more efficient and controllable manufacturing protocols. The process begins with the preparation of the azide-functionalized linker backbone, followed by a series of protection and deprotection steps that build the necessary functionality for payload attachment. A critical aspect of this workflow is the isolation of the stable carboxylic acid intermediate, which serves as a quality gate before the introduction of the cytotoxic agent. This modular design allows for the stockpiling of high-quality linker precursors, decoupling the supply of the linker from the availability of the payload. The detailed standardized synthesis steps involve precise stoichiometric control, specific solvent systems like THF and DMF, and careful temperature management to ensure reproducibility. For a comprehensive understanding of the operational parameters, including exact reagent quantities and workup procedures, the detailed standardized synthesis steps are provided in the guide below.
- Synthesize Formula VI compound via substitution reaction with paraformaldehyde and trimethylsilyl chloride.
- Perform etherification with Reagent V to obtain Formula IV compound, followed by azide reduction to Formula III.
- Couple Formula III with EMCS to get Formula II, deprotect to Formula I, and finally conjugate with Exatecan mesylate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers transformative benefits that extend beyond mere technical superiority. The primary advantage lies in the significant optimization of raw material utilization, particularly regarding the cytotoxic payload. By postponing the introduction of Exatecan to the final step, the process eliminates the financial risk associated with losing expensive drug substances during early-stage purification failures. This strategic delay means that the high-value payload is only consumed when the linker is fully verified and qualified, drastically reducing the cost of goods sold (COGS). Furthermore, the stability of the key intermediates allows for larger batch sizes and longer storage times, enhancing inventory flexibility and reducing the pressure on just-in-time manufacturing schedules. This robustness translates directly into a more resilient supply chain capable of withstanding market fluctuations and demand surges without compromising on delivery timelines.
- Cost Reduction in Manufacturing: The elimination of unstable intermediates that previously required immediate processing reduces waste generation and solvent consumption. By avoiding the use of expensive DXd derivatives in favor of direct Exatecan conjugation at the end, the material costs are substantially lowered. The ability to purify intermediates effectively means less resource expenditure on remedial purification of the final product, leading to overall process efficiency gains.
- Enhanced Supply Chain Reliability: The modular nature of this synthesis allows for the independent production and quality control of the linker component. This decoupling mitigates the risk of supply disruptions caused by payload shortages. Additionally, the use of commercially available and stable reagents for the linker backbone ensures a consistent supply of starting materials, reducing lead time for high-purity linker drug conjugates and enabling more accurate forecasting for production planning.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up of complex pharmaceutical intermediates in mind, utilizing standard unit operations like extraction and column chromatography that are easily transferable from pilot to production scale. The reduced use of hazardous reagents and the minimization of cytotoxic exposure align with modern environmental, health, and safety (EHS) standards, lowering the regulatory burden and facility containment costs associated with handling potent compounds.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation. Understanding these nuances is critical for stakeholders evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The answers reflect the specific advantages of the stable intermediate strategy and the quality outcomes observed in pilot studies.
Q: Why is the new synthesis route for LE14 more cost-effective than prior art?
A: The new route avoids using expensive DXd or DXd derivatives in early steps, introducing the cytotoxic payload only at the final stage, which significantly reduces material loss and handling costs.
Q: How does the new method improve intermediate stability?
A: Unlike prior routes where carboxylic acid intermediates were unstable and decomposed during purification, the key intermediate I in this patent is stable, allowing for effective purification and higher overall purity.
Q: What represents the critical quality attribute for LE14 in this process?
A: The process ensures the final LE14 product meets IND declaration requirements with total impurities not exceeding 3% and single impurities not exceeding 1%, achieved through stable intermediate handling.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable LE14 Supplier
As the demand for next-generation ADC therapies continues to surge, partnering with a manufacturer who possesses deep technical expertise in linker-payload conjugation is essential. NINGBO INNO PHARMCHEM stands at the forefront of this industry, leveraging advanced synthetic methodologies like the one described in CN116217654A to deliver superior products. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from development to market. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of LE14 meets the highest global standards. Our commitment to quality assurance means that we can consistently provide materials that satisfy the demanding requirements of IND filings and clinical trials.
We invite you to explore how our optimized manufacturing capabilities can enhance your drug development pipeline. By collaborating with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume needs and timeline constraints. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments for your upcoming projects. Let us help you navigate the complexities of ADC manufacturing with confidence, ensuring a secure and efficient supply of critical intermediates for your life-saving therapies.