Scalable Manufacturing of High-Purity Cefdinir via Novel Silanization and Salt Formation Route
Introduction to Advanced Cefdinir Manufacturing Technology
The global demand for third-generation cephalosporin antibiotics continues to rise, driven by the need for effective treatments against resistant bacterial strains in respiratory and soft tissue infections. At the forefront of this therapeutic class is Cefdinir, a potent semi-synthetic antibiotic whose commercial viability relies heavily on the efficiency and purity of its manufacturing process. Patent CN102010427B introduces a transformative methodology that addresses long-standing challenges in Cefdinir synthesis, specifically focusing on the optimization of the acylation and purification stages. This technical insight report analyzes the proprietary route which utilizes a strategic silanization protection of the 7-amino-3-vinyl-3-cephem-4-carboxylic acid (7-AVCA) nucleus, followed by a unique salt-formation purification step. By shifting away from hazardous acyl chloride intermediates and cumbersome trityl protection groups, this process offers a robust pathway for producing high-purity Cefdinir suitable for direct clinical use.
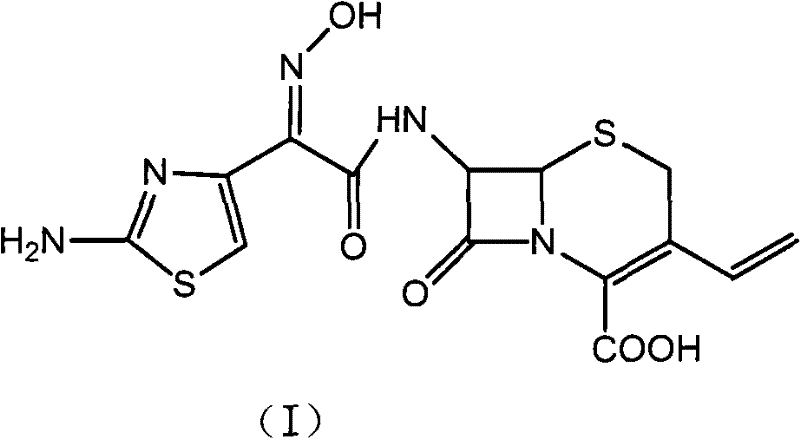
The significance of this patent lies in its ability to harmonize chemical selectivity with industrial practicality. Traditional methods often struggle with the stability of the beta-lactam ring and the stereochemical integrity of the oxime side chain. The disclosed method mitigates these risks through controlled reaction conditions ranging from 0°C to 50°C, utilizing common organic solvents such as methylene dichloride and ethyl acetate. For pharmaceutical manufacturers and procurement specialists, understanding the mechanistic advantages of this route is essential for securing a reliable supply chain of high-quality antibiotic intermediates. The following sections provide a deep dive into the comparative advantages, mechanistic insights, and commercial implications of adopting this silanization-based synthesis strategy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Cefdinir has been plagued by several distinct methodologies, each carrying significant drawbacks that impact yield, safety, and final product quality. The chloride method, described in early Japanese patents, involves the conversion of the side chain acid to an acyl chloride prior to coupling. While chemically straightforward, this approach suffers from severe security risks associated with handling reactive acyl chlorides on a large scale. Furthermore, the chloride method frequently fails to secure the Z-isomer configuration with the necessary specificity, often resulting in e.e. values below the required 99%, which necessitates complex and yield-reducing purification steps. Another prevalent technique involves the use of active esters, such as mercaptobenzothiazole derivatives. Although safer than acyl chlorides, direct acylation using these esters in mixed solvent systems often results in crude products with lower content and purity, requiring extensive downstream refining that erodes overall process efficiency.
Additionally, alternative routes utilizing trityl protecting groups for the oxime hydroxyl function have been explored to improve condensation smoothness. However, the removal of the bulky trityl group is a time-consuming process, often requiring several hours of acidolysis. This extended reaction time not only lowers the throughput of the manufacturing facility but also increases the exposure of the sensitive beta-lactam core to acidic conditions, potentially leading to degradation and increased impurity profiles. These conventional limitations create a bottleneck for suppliers aiming to deliver cost-effective, high-purity Cefdinir to the global market, highlighting the urgent need for a more streamlined and selective synthetic approach.
The Novel Approach
The methodology outlined in Patent CN102010427B represents a significant departure from these legacy processes by introducing a dual-protection and salt-crystallization strategy. The core innovation begins with the silanization of the carboxyl group on the 7-AVCA starting material. By converting the carboxylic acid into a silyl ester using reagents like BSA (N,O-bis(trimethylsilyl)acetamide), the process effectively masks the C4 functionality, preventing it from interfering during the critical C7 acylation step. This ensures that the subsequent reaction with the Cefdinir Active Ester (CAEM) proceeds with high regioselectivity. Following acylation, the process does not immediately attempt to deprotect the oxime group. Instead, it employs a clever intermediate isolation technique where the acetyl-protected cefdinir is converted into a dicyclohexylamine salt.
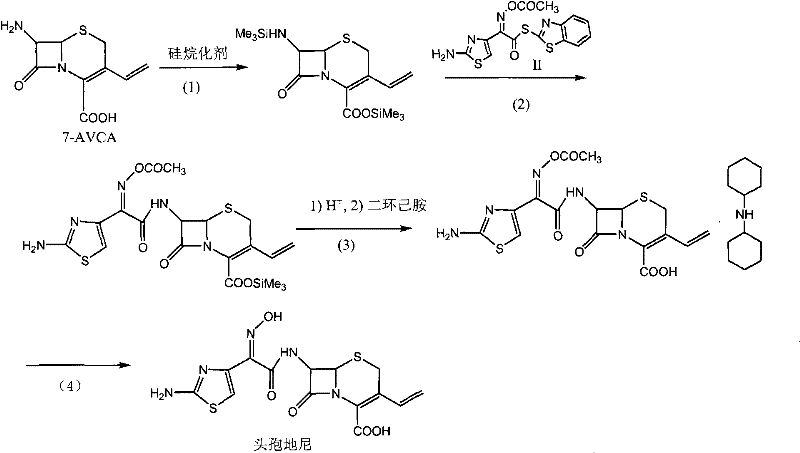
This salt formation step is the linchpin of the novel approach. By precipitating the intermediate as a dicyclohexylamine salt, the process achieves a high degree of purification before the final hydrolysis. Impurities and unreacted starting materials remain in the mother liquor, while the desired intermediate crystallizes out as a white solid with exceptional purity. This pre-purification allows the final hydrolysis step to proceed on a already refined substrate, drastically reducing the burden on the final crystallization of the API. The result is a process that is not only simpler and quicker, with reaction times measured in minutes for the salt formation, but also delivers a product that can be directly used as a bulk drug for clinical applications without further intensive refining.
Mechanistic Insights into Silanization and Selective Acylation
To fully appreciate the technical superiority of this route, one must examine the electronic and steric factors governing the silanization and acylation sequence. The 7-AVCA molecule possesses two nucleophilic sites: the primary amine at the C7 position and the carboxylic acid at the C4 position. In traditional acylation without protection, competitive reactions can occur, or the carboxylic acid can catalyze unwanted degradation pathways. The introduction of the trimethylsilyl group transforms the carboxylic acid into a silyl ester, which is stable under the mild acylation conditions (20-25°C) used in the second step. This temporary protection ensures that the nucleophilic attack of the C7 amine on the carbonyl carbon of the CAEM active ester is the dominant reaction pathway. The use of CAEM, which features an acetyl-protected oxime, further stabilizes the side chain, preventing isomerization from the biologically active Z-form to the inactive E-form during the coupling reaction.
Furthermore, the mechanism of the salt formation step leverages the acid-base properties of the intermediate. The acetyl cefdinir intermediate retains the carboxylic acid functionality (after the silyl group is removed during the workup or concurrently). When treated with dicyclohexylamine in a solvent system comprising acetone and methanol, a stable ammonium carboxylate salt is formed. 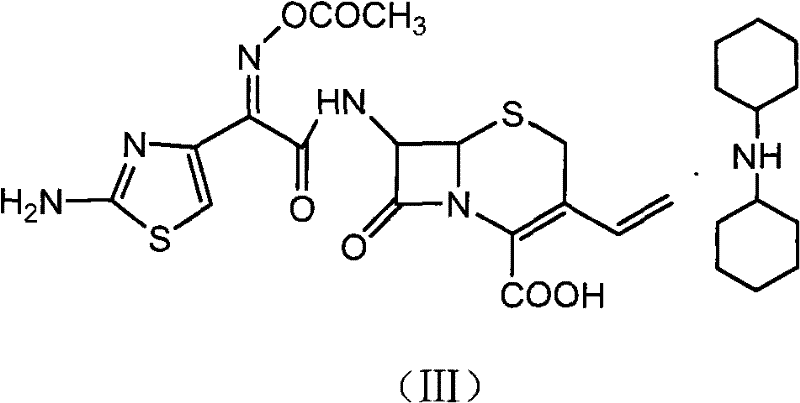 The crystal lattice energy of this specific salt is high enough to drive precipitation from the solution, even in the presence of potential impurities. This phenomenon is critical for impurity control; it acts as a 'chemical filter' that excludes structurally similar by-products which do not fit into the crystal lattice or remain soluble in the cold solvent mixture. The subsequent hydrolysis of this purified salt under controlled acidic or alkaline conditions then cleanly removes the acetyl group from the oxime, revealing the final hydroxyl functionality of Cefdinir without compromising the integrity of the beta-lactam ring.
The crystal lattice energy of this specific salt is high enough to drive precipitation from the solution, even in the presence of potential impurities. This phenomenon is critical for impurity control; it acts as a 'chemical filter' that excludes structurally similar by-products which do not fit into the crystal lattice or remain soluble in the cold solvent mixture. The subsequent hydrolysis of this purified salt under controlled acidic or alkaline conditions then cleanly removes the acetyl group from the oxime, revealing the final hydroxyl functionality of Cefdinir without compromising the integrity of the beta-lactam ring.
How to Synthesize Cefdinir Efficiently
The implementation of this synthesis route requires precise control over stoichiometry and temperature to maximize the benefits of the silanization and salt-formation steps. The process is designed to be telescoped where possible, minimizing the isolation of unstable intermediates. Operators must ensure that the silanization reaction proceeds to completion, indicated by the clear dissolution of the 7-AVCA starting material, before introducing the active ester. The subsequent acylation is monitored via HPLC to ensure residual 7-AVCA levels are minimized (<3mg/ml) before proceeding to the salt formation. The cooling profile during the salt precipitation is also vital; rapid cooling to 0°C followed by stirring ensures the formation of fine, pure crystals. For a detailed breakdown of the specific operational parameters, reagent ratios, and workup procedures, please refer to the standardized synthesis guide below.
- Perform silanization protection on the carboxyl group of 7-AVCA using a silylating reagent in an organic solvent at 0-50°C.
- Conduct acylation reaction by adding CAEM (2-(thiazolamine-4-yl)-2-(Z)-(acetyl oxygen imido group) acetyl benzothiazole thioesters) to the silanized solution.
- Form the acetyl cefdinir dicyclohexylamine salt through hydrolysis or alcoholysis followed by reaction with dicyclohexylamine.
- Hydrolyze the salt under acidic or alkaline conditions to remove the acetyl protecting group and isolate pure Cefdinir.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible benefits that extend beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the purification train. By integrating a high-efficiency crystallization step via the dicyclohexylamine salt, the need for multiple chromatographic separations or repetitive recrystallizations is eliminated. This reduction in unit operations translates directly into lower manufacturing costs, as it reduces solvent consumption, energy usage for heating and cooling cycles, and labor hours required for processing. The ability to produce a product suitable for direct clinical use implies a higher first-pass yield, meaning less raw material is wasted on re-processing off-spec batches, thereby optimizing the cost of goods sold (COGS) for the final API.
- Cost Reduction in Manufacturing: The elimination of hazardous acyl chlorides removes the need for specialized corrosion-resistant equipment and expensive safety protocols associated with handling reactive acid chlorides. Furthermore, the use of common, commodity solvents such as ethyl acetate, acetone, and methylene dichloride ensures that raw material costs remain stable and predictable. The high yield reported in the patent embodiments (over 90% for the salt formation and final hydrolysis steps) indicates a highly atom-economical process that minimizes waste disposal costs. By avoiding the lengthy deprotection times associated with trityl groups, reactor occupancy time is significantly reduced, allowing for greater throughput within existing infrastructure.
- Enhanced Supply Chain Reliability: The reliance on 7-AVCA and CAEM as starting materials leverages a well-established supply chain for cephalosporin intermediates. These key building blocks are widely produced by specialty chemical manufacturers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions, which tolerate a range of temperatures between 0°C and 50°C, makes the process less susceptible to minor fluctuations in plant utilities, ensuring consistent batch-to-batch quality. This reliability is crucial for maintaining continuous supply to downstream formulation partners who depend on just-in-time delivery of high-quality antibiotic substances.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively from gram to multi-kilogram scales in the patent examples. The absence of heavy metal catalysts or exotic reagents simplifies the environmental compliance profile, making it easier to obtain necessary regulatory approvals for production facilities. The waste streams generated are primarily organic solvents and aqueous salts, which can be managed through standard recovery and treatment systems. This alignment with green chemistry principles not only reduces the environmental footprint but also future-proofs the manufacturing site against increasingly stringent global environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Cefdinir synthesis route. These answers are derived directly from the experimental data and claims presented in Patent CN102010427B, providing clarity on the operational feasibility and quality outcomes of the process. Understanding these details is vital for technical teams evaluating the transfer of this technology to commercial production scales.
Q: How does the silanization step improve the purity of Cefdinir?
A: The silanization step protects the carboxyl group at the C4 position of the 7-AVCA nucleus. This prevents unwanted side reactions during the subsequent acylation at the C7 position, significantly reducing impurity formation and improving the selectivity of the acetyl cefdinir intermediate.
Q: Why is the dicyclohexylamine salt formation critical in this process?
A: Forming the dicyclohexylamine salt allows for the crystallization and isolation of the acetyl cefdinir intermediate in a highly pure solid form. This step effectively separates the product from reaction by-products and unreacted starting materials before the final deprotection, ensuring the final API meets stringent clinical standards.
Q: What are the advantages of this method over the traditional chloride method?
A: Unlike the chloride method which poses significant safety risks due to unstable acyl chlorides and often fails to achieve high Z-isomer purity (>99%), this silanization-based route operates under milder conditions. It avoids hazardous reagents, simplifies the workflow, and consistently yields high-purity product suitable for direct clinical application.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cefdinir Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team specializes in the scale-up of complex antibiotic intermediates, including the silanization-based synthesis of Cefdinir described in this report. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high purity and yield observed in the lab are faithfully reproduced in our pilot and commercial plants. Our state-of-the-art facilities are equipped with rigorous QC labs capable of monitoring critical parameters such as Z-isomer content and residual solvent levels, guaranteeing that every batch meets stringent purity specifications required by global pharmacopoeias.
We invite pharmaceutical companies and generic drug manufacturers to collaborate with us to optimize their Cefdinir supply chain. By leveraging our advanced process chemistry capabilities, we can help you achieve significant cost reductions and supply security. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing excellence can support your product lifecycle goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →