Scalable Synthesis of Trifluoromethyl 1,2,4-Triazines for Advanced Pharmaceutical Applications
Scalable Synthesis of Trifluoromethyl 1,2,4-Triazines for Advanced Pharmaceutical Applications
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting potent biological activity. Patent CN116253692A introduces a groundbreaking preparation method for trifluoromethyl substituted 1,2,4-triazine compounds, addressing critical bottlenecks in the production of high-value pharmaceutical intermediates. These heterocyclic structures are ubiquitous in drug discovery, serving as key motifs in agents with anticancer, antifungal, anti-inflammatory, and antimalarial properties, as illustrated by the diverse bioactive molecules shown in the reference data. The strategic incorporation of trifluoromethyl groups into these scaffolds significantly enhances physicochemical properties such as metabolic stability, lipophilicity, and bioavailability, making them indispensable for next-generation therapeutics. This patent discloses a novel pathway that leverages cheap and easily accessible starting materials, specifically chlorohydrazones and trifluoroacetyl thio ylides, to construct these complex cores with remarkable efficiency.
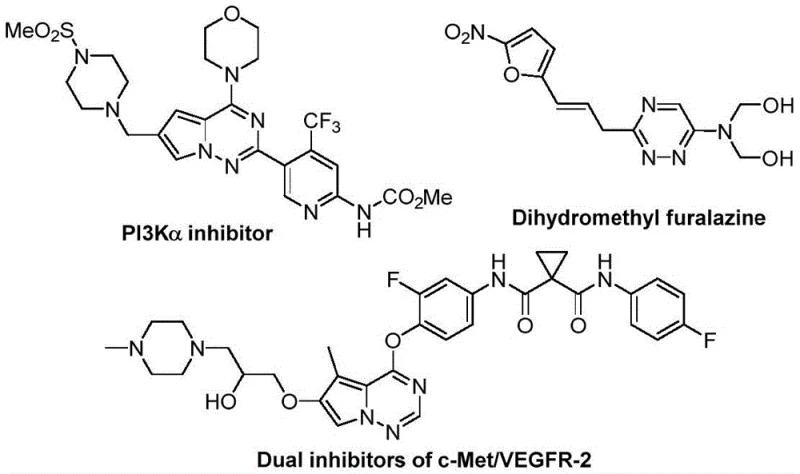
For R&D directors and process chemists, the significance of this invention lies in its ability to bypass the limitations of conventional synthesis strategies. Traditional methods for constructing 1,2,4-triazine rings often rely on the condensation of amidrazones with 1,2-diketones or alkynes, or multicomponent reactions involving hydrazides and dicarbonyl compounds. While effective in specific contexts, these legacy approaches frequently suffer from harsh reaction conditions, poor atom economy, and limited structural diversity, which can severely hamper the rapid exploration of chemical space during lead optimization phases. Furthermore, the specific introduction of trifluoromethyl groups using older methodologies often requires specialized reagents or multi-step sequences that reduce overall yield and increase waste generation. The novel approach detailed in this patent circumvents these issues by utilizing a synergistic [3+3] cycloaddition strategy that is both operationally simple and chemically robust.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 1,2,4-triazine derivatives has been plagued by several intrinsic drawbacks that impact both research throughput and manufacturing viability. Conventional cyclization reactions often necessitate the use of strong acids or bases, elevated temperatures, and inert atmospheres, which increases energy consumption and operational complexity. Additionally, many traditional routes involve the use of transition metal catalysts, which not only add to the raw material cost but also introduce the risk of heavy metal contamination in the final active pharmaceutical ingredient (API). Removing trace metals to meet stringent regulatory standards requires additional purification steps, such as scavenging or recrystallization, which inevitably lowers the overall process yield. Moreover, the substrate scope in classical methods is often narrow, limiting the ability of chemists to introduce diverse functional groups at specific positions on the triazine ring, thereby restricting the SAR (Structure-Activity Relationship) studies essential for drug development.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN116253692A offers a streamlined and environmentally benign alternative that transforms the synthesis landscape for these valuable intermediates. The core innovation involves the reaction of a chlorohydrazone derivative with a trifluoroacetyl sulfur ylide in the presence of potassium carbonate as a promoter. This reaction proceeds smoothly at room temperature (20-40°C) in an air atmosphere, eliminating the need for expensive inert gas protection or energy-intensive heating. The general reaction scheme highlights the elegance of this transformation, where a nitrile imine intermediate is generated in situ and subsequently undergoes cycloaddition with the sulfur ylide. This metal-free protocol not only simplifies the workup procedure—often requiring only filtration and chromatography—but also ensures that the final product is free from toxic metal residues, a critical advantage for pharmaceutical applications.
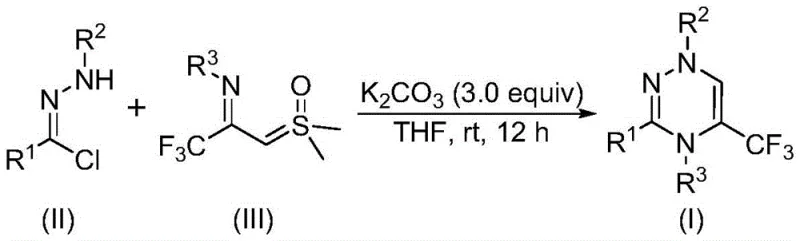
Mechanistic Insights into Metal-Free [3+3] Cycloaddition
From a mechanistic perspective, this transformation represents a sophisticated interplay of nucleophilic and electrophilic species driven by the basicity of potassium carbonate. The reaction initiates with the deprotonation of the chlorohydrazone by the carbonate base, leading to the elimination of hydrogen chloride and the formation of a highly reactive nitrile imine intermediate. This 1,3-dipole then engages in a concerted [3+3] cycloaddition with the electron-deficient carbon-carbon double bond of the trifluoroacetyl sulfur ylide. The driving force for this cyclization is further augmented by the subsequent elimination of dimethyl sulfoxide (DMSO), which acts as a leaving group to aromatize the system and form the stable 1,2,4-triazine ring. This mechanism is particularly advantageous because it avoids the formation of stable organometallic complexes that can be difficult to decompose, thereby reducing the formation of intractable byproducts and simplifying the impurity profile of the crude reaction mixture.
The control of impurities in this process is inherently superior due to the mild reaction conditions and the specificity of the cycloaddition. Unlike radical-based or high-temperature thermal cyclizations that can lead to polymerization or decomposition of sensitive functional groups, this room-temperature protocol preserves the integrity of substituents such as halogens, alkoxy groups, and trifluoromethyl moieties. The use of tetrahydrofuran (THF) as the preferred solvent ensures excellent solubility of both organic reactants and the inorganic base, facilitating homogeneous reaction kinetics that minimize localized hot spots or concentration gradients which often lead to side reactions. Consequently, the resulting trifluoromethyl substituted 1,2,4-triazine compounds, such as the diverse array of derivatives shown in the specific examples, are obtained with high purity and excellent yields, demonstrating the robustness of this synthetic design across a broad substrate scope.
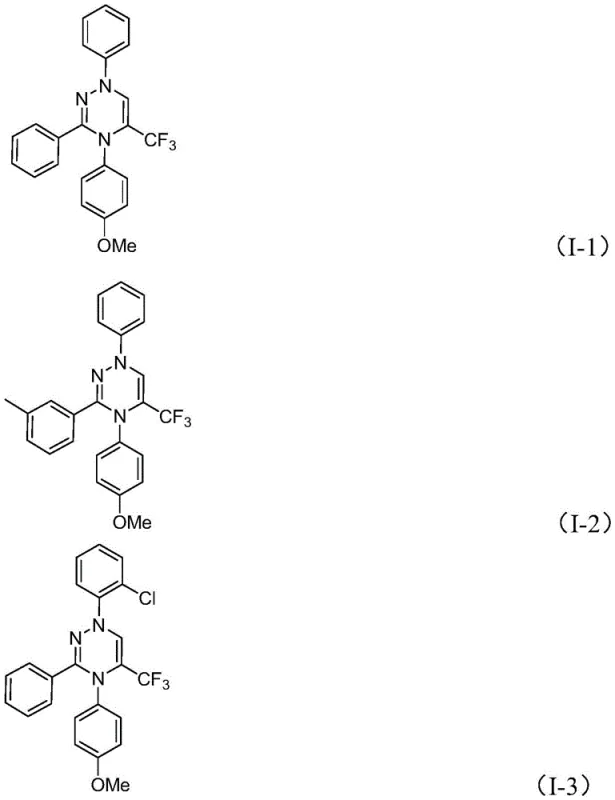
How to Synthesize Trifluoromethyl Substituted 1,2,4-Triazine Efficiently
The practical execution of this synthesis is designed for ease of operation, making it accessible for both laboratory-scale discovery and pilot-scale production. The process begins by simply combining the chlorohydrazone, trifluoroacetyl sulfur ylide, and potassium carbonate in an organic solvent, typically THF, within a standard reaction vessel. The mixture is stirred at ambient temperature for a period of 10 to 14 hours, allowing the reaction to reach completion without the need for rigorous monitoring or complex control systems. Following the reaction, the workup involves a straightforward filtration to remove inorganic salts, followed by standard purification techniques like column chromatography to isolate the target molecule. For a detailed breakdown of the specific stoichiometric ratios, solvent volumes, and purification parameters validated in the patent examples, please refer to the standardized synthesis guide below.
- Combine potassium carbonate, chlorohydrazone, and trifluoroacetyl sulfur ylide in an organic solvent such as tetrahydrofuran.
- Stir the reaction mixture at room temperature (20-40°C) in an air atmosphere for 10 to 14 hours to ensure complete conversion.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the high-purity trifluoromethyl substituted 1,2,4-triazine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this synthetic route offers compelling economic and logistical benefits that directly impact the bottom line. The elimination of heavy metal catalysts removes a significant cost center associated with both the purchase of precious metals and the downstream processing required to meet residual metal specifications. Furthermore, the reliance on potassium carbonate, an inexpensive and non-toxic inorganic salt, drastically reduces the raw material expenditure compared to processes requiring exotic organometallic reagents or strong, hazardous bases. The ability to run the reaction in air at room temperature also translates to substantial energy savings, as there is no requirement for cryogenic cooling, high-pressure reactors, or nitrogen blanketing systems, thereby lowering the overall utility costs of manufacturing.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of commodity chemicals that are readily available in the global market. By avoiding the need for specialized, high-cost catalysts and reducing the number of unit operations required for purification, manufacturers can achieve a leaner cost structure. The high atom economy of the cycloaddition reaction ensures that a greater proportion of the input mass is converted into the desired product, minimizing waste disposal costs and maximizing material efficiency. This streamlined approach allows for a more competitive pricing strategy for the final trifluoromethyl 1,2,4-triazine intermediates, providing a distinct advantage in cost-sensitive pharmaceutical supply chains.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the accessibility of the starting materials, namely chlorohydrazones and trifluoroacetyl sulfur ylides, which can be sourced from multiple vendors or synthesized via simple, well-established protocols. The robustness of the reaction conditions means that production is less susceptible to disruptions caused by equipment failure or environmental fluctuations, ensuring consistent output quality and volume. Additionally, the absence of air- or moisture-sensitive reagents simplifies logistics and storage requirements, reducing the risk of material degradation during transport and allowing for more flexible inventory management strategies across global distribution networks.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this metal-free methodology aligns perfectly with the industry's shift towards greener chemistry practices. The avoidance of toxic heavy metals simplifies the regulatory approval process for new drug applications by reducing the burden of extensive metal testing and validation. The process generates minimal hazardous waste, primarily consisting of benign inorganic salts and organic solvents that can be readily recovered and recycled. This eco-friendly profile not only reduces the environmental footprint of manufacturing but also mitigates compliance risks, making it an ideal candidate for sustainable commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
To address common inquiries regarding the technical implementation and commercial potential of this technology, we have compiled a set of answers based on the detailed experimental data and mechanistic insights provided in the patent literature. These questions cover critical aspects ranging from reaction optimization to substrate compatibility, offering clarity for technical teams evaluating this route for their specific projects. Understanding these nuances is essential for effectively integrating this methodology into existing production workflows and maximizing its value proposition.
Q: What are the primary advantages of this synthesis method over traditional routes?
A: This method eliminates the need for expensive and toxic heavy metal catalysts, operates under mild room temperature conditions in air, and utilizes cheap, readily available inorganic salts like potassium carbonate as promoters.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process has been demonstrated to be scalable to gram levels with simple post-treatment procedures like filtration and column chromatography, making it highly amenable to industrial scale-up.
Q: What is the structural diversity achievable with this synthetic route?
A: The method supports a wide range of substituents including alkyl, phenyl, naphthyl, and furyl groups with various functional groups like halogens and alkoxy groups, allowing for the design of diverse heterocyclic libraries.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl 1,2,4-Triazine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this metal-free synthesis for the production of high-purity pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to full-scale manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of trifluoromethyl substituted 1,2,4-triazine compound delivered meets the highest international standards for safety and efficacy. We are committed to leveraging our technical expertise to optimize this novel route for your specific application needs.
We invite you to collaborate with us to explore the full capabilities of this advanced synthetic technology. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your project requirements, demonstrating how this efficient route can enhance your supply chain performance. Please contact us today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your custom synthesis projects, and let us help you accelerate your drug development timeline with reliable, high-quality intermediates.