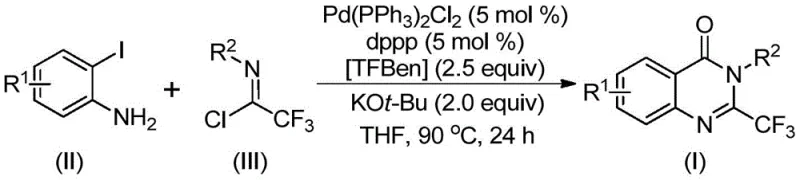
Advanced Palladium-Catalyzed Carbonylation for Scalable 2-Trifluoromethyl Quinazolinone Manufacturing
Advanced Palladium-Catalyzed Carbonylation for Scalable 2-Trifluoromethyl Quinazolinone Manufacturing
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing nitrogen-containing heterocycles, particularly quinazolinone derivatives, which serve as critical scaffolds in numerous bioactive molecules ranging from anticancer agents to antiviral drugs. A significant technological breakthrough in this domain is documented in patent CN112125856A, which discloses a novel preparation method for 2-trifluoromethyl substituted quinazolinone derivatives. This innovation addresses long-standing challenges in heterocyclic synthesis by employing a transition metal palladium-catalyzed carbonylation tandem reaction that utilizes inexpensive and readily available starting materials. The introduction of the trifluoromethyl group is strategically vital, as it significantly enhances the metabolic stability, lipophilicity, and bioavailability of the parent drug molecules, making this synthetic route highly valuable for the development of next-generation therapeutic agents. By leveraging a solid carbon monoxide substitute, this process not only improves operational safety but also streamlines the manufacturing workflow, positioning it as a superior alternative for reliable pharmaceutical intermediate supplier networks aiming to optimize their production portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinone derivatives has been fraught with significant technical and safety hurdles that impede efficient large-scale manufacturing. Conventional literature reports often rely on the direct use of carbon monoxide gas, a highly toxic and colorless substance that necessitates specialized high-pressure equipment and rigorous safety protocols to prevent lethal exposure incidents. Alternative methods involving the cyclization of anthranilamides with ethyl trifluoroacetate or trifluoroacetic anhydride frequently suffer from harsh reaction conditions, requiring extreme temperatures or strong acidic environments that can degrade sensitive functional groups on the substrate. Furthermore, many traditional pathways involve expensive pre-activated substrates or unstable intermediates like trifluoroacetamides, which complicate the supply chain and drive up the cost of goods sold due to low atom economy and poor overall yields. These limitations collectively restrict the substrate scope, making it difficult to introduce diverse substituents without compromising the integrity of the final product, thereby limiting the chemical space available for medicinal chemists during drug discovery phases.
The Novel Approach
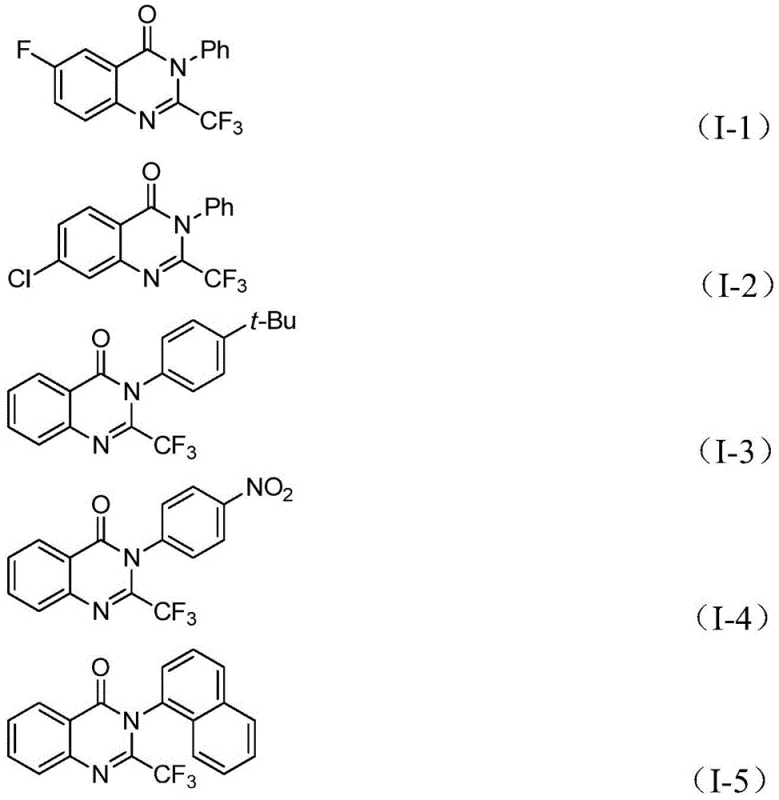
In stark contrast to these legacy techniques, the methodology described in patent CN112125856A introduces a paradigm shift by utilizing 1,3,5-tricarboxylate phenol ester (TFBen) as a safe, solid carbon monoxide substitute. This innovative approach allows for the generation of carbon monoxide in situ under mild thermal conditions, effectively bypassing the need for handling hazardous gas cylinders while maintaining high reaction efficiency. The process employs a palladium catalyst system, specifically Pd(PPh3)2Cl2 paired with the bidentate ligand 1,3-bis(diphenylphosphine)propane (dppp), which facilitates a smooth carbonylation cyclization between o-iodoaniline and trifluoroethylimidoyl chloride. This catalytic system exhibits remarkable tolerance for a wide array of functional groups, including halogens, alkyl chains, and nitro groups, allowing for the synthesis of a diverse library of derivatives without the need for extensive protecting group strategies. The operational simplicity, combined with the use of cheap and commercially available raw materials, makes this route exceptionally attractive for cost reduction in pharmaceutical intermediate manufacturing, offering a scalable solution that aligns with modern green chemistry principles.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The mechanistic pathway of this transformation involves a sophisticated sequence of organometallic steps initiated by the oxidative addition of the palladium catalyst into the carbon-iodine bond of the o-iodoaniline substrate. Following this activation, the solid CO surrogate TFBen decomposes under the reaction temperature of 90°C to release carbon monoxide, which subsequently inserts into the carbon-palladium bond to form a reactive acyl-palladium intermediate. This insertion step is critical for introducing the carbonyl functionality that becomes part of the quinazolinone ring system. Concurrently, the trifluoroethylimidoyl chloride interacts with the base, potassium tert-butoxide, to facilitate the formation of a trifluoroacetamidine species, which then coordinates with the palladium center. The subsequent intramolecular nucleophilic attack and reductive elimination steps close the ring, releasing the final 2-trifluoromethyl substituted quinazolinone product and regenerating the active palladium catalyst for the next cycle. This intricate dance of ligands and substrates ensures high selectivity and minimizes the formation of side products, which is essential for maintaining high purity standards required in pharmaceutical applications.
From an impurity control perspective, the choice of the dppp ligand plays a pivotal role in stabilizing the palladium intermediates and preventing the formation of homocoupling byproducts that often plague cross-coupling reactions. The use of THF as the solvent further optimizes the solubility of all reactants and intermediates, ensuring a homogeneous reaction environment that promotes consistent kinetics throughout the batch. The base, potassium tert-butoxide, not only deprotonates the amine but also assists in the neutralization of hydrochloric acid generated during the reaction, preventing catalyst poisoning. By carefully balancing the molar ratios of the catalyst, ligand, and base, the process achieves high conversion rates even with sterically hindered substrates, demonstrating the robustness of the catalytic cycle. This deep understanding of the mechanism allows process chemists to fine-tune reaction parameters to maximize yield and minimize waste, directly impacting the economic viability of the synthesis.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The practical execution of this synthesis requires precise adherence to the optimized conditions outlined in the patent to ensure reproducibility and high quality outcomes. The process begins with the careful weighing and mixing of the palladium catalyst, ligand, base, solid CO source, and the two primary organic substrates in an inert atmosphere to prevent catalyst oxidation. The reaction mixture is then heated to a controlled temperature of 90°C and maintained for a period ranging from 16 to 30 hours, depending on the specific electronic nature of the substituents on the aromatic rings. Detailed standardized synthetic steps for this procedure are provided in the guide below, which outlines the exact stoichiometry and workup procedures necessary to isolate the pure product.
- Combine palladium catalyst Pd(PPh3)2Cl2, ligand dppp, base KOt-Bu, solid CO source TFBen, trifluoroethylimidoyl chloride, and o-iodoaniline in an organic solvent like THF.
- Heat the reaction mixture to 90°C and maintain stirring for 16 to 30 hours to allow the carbonylation cyclization to proceed to completion.
- Upon completion, filter the mixture, mix with silica gel, and purify the crude product via column chromatography to isolate the target 2-trifluoromethyl quinazolinone derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers substantial strategic benefits that extend beyond mere chemical efficiency. The elimination of toxic carbon monoxide gas from the process significantly reduces the regulatory burden and infrastructure costs associated with hazardous material storage and handling, leading to a safer working environment and lower insurance premiums. Furthermore, the reliance on commercially available and inexpensive starting materials such as o-iodoanilines and trifluoroethylimidoyl chlorides ensures a stable and resilient supply chain that is less susceptible to market volatility or geopolitical disruptions. The broad substrate scope of this method means that a single manufacturing platform can be adapted to produce a wide variety of derivatives, enhancing asset utilization and reducing the need for dedicated production lines for each specific compound. This flexibility allows for rapid response to changing market demands and accelerates the timeline from clinical trials to commercial launch.
- Cost Reduction in Manufacturing: The use of a solid carbon monoxide surrogate eliminates the need for expensive high-pressure reactors and specialized gas handling equipment, resulting in significant capital expenditure savings. Additionally, the high atom economy and excellent yields reported in the patent minimize raw material waste, directly lowering the variable cost per kilogram of the final API intermediate. The avoidance of complex protection and deprotection steps further streamlines the synthesis, reducing labor hours and utility consumption associated with prolonged reaction times and multiple purification stages.
- Enhanced Supply Chain Reliability: Since the key raw materials are commodity chemicals widely produced by the global chemical industry, the risk of supply shortages is markedly reduced compared to methods relying on bespoke or unstable reagents. The robustness of the catalytic system ensures consistent batch-to-batch quality, which is critical for maintaining regulatory compliance and avoiding costly production delays due to out-of-specification results. This reliability fosters stronger partnerships between manufacturers and their downstream pharmaceutical clients, ensuring uninterrupted delivery of critical drug substances.
- Scalability and Environmental Compliance: The homogeneous nature of the reaction in THF solvent facilitates straightforward scale-up from laboratory to pilot and eventually to commercial production scales without encountering significant heat transfer or mixing issues. The simplified post-treatment process, involving filtration and standard column chromatography, generates less hazardous waste compared to traditional methods, aligning with increasingly stringent environmental regulations. This eco-friendly profile enhances the corporate sustainability image and reduces the costs associated with waste disposal and environmental remediation.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this palladium-catalyzed carbonylation process. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, providing clarity on reaction scope and operational parameters. Understanding these nuances is essential for process engineers and quality assurance teams tasked with validating the method for GMP manufacturing environments.
Q: What is the primary safety advantage of this synthesis method compared to traditional carbonylation?
A: This method utilizes 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute, effectively eliminating the need for handling toxic and hazardous carbon monoxide gas directly.
Q: Does this catalytic system support a wide range of substrate substituents?
A: Yes, the protocol demonstrates excellent compatibility with various substituents on both the o-iodoaniline and the imidoyl chloride, including halogens, alkyl groups, and nitro groups, ensuring broad applicability.
Q: What are the typical reaction conditions required for this transformation?
A: The reaction typically proceeds in an aprotic solvent such as THF at a temperature of 90°C for a duration of 16 to 30 hours, utilizing a palladium catalyst and potassium tert-butoxide as the base.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting cutting-edge synthetic technologies to meet the evolving needs of the global pharmaceutical industry. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to full-scale manufacturing is seamless and efficient. We are committed to delivering high-purity 2-trifluoromethyl quinazolinone derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our dedication to quality and safety makes us a trusted partner for companies seeking to secure a stable supply of complex heterocyclic intermediates.
We invite you to collaborate with us to leverage this advanced synthetic route for your specific drug development projects. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your project timelines and optimize your overall budget.