Advanced Catalytic Synthesis of 2-Trifluoromethyl Imidazoles: From Lab to Commercial Scale
Patent CN111423381A introduces a novel palladium-catalyzed carbonylation methodology for synthesizing 2-trifluoromethyl substituted imidazole compounds, a critical structural motif in pharmaceutical development. This breakthrough process addresses longstanding challenges in producing high-purity heterocyclic compounds with trifluoromethyl groups, which significantly enhance drug candidates' bioavailability and metabolic stability as documented in J.Med.Chem.2015,58,8315-8359. The methodology leverages readily available starting materials including trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salts under mild conditions (30°C, 18–24 hours) to deliver structurally diverse imidazole intermediates essential for modern drug discovery pipelines.
Overcoming Traditional Limitations in Trifluoromethyl Imidazole Synthesis
The Limitations of Conventional Methods
Traditional approaches to synthesizing trifluoromethylated imidazoles often rely on harsh reaction conditions or expensive trifluoromethylation reagents that compromise both yield and purity. Conventional methods using trifluorodiazoethane require cryogenic temperatures and generate hazardous byproducts, while alternative routes involving transition metal catalysts frequently suffer from poor functional group tolerance and extensive purification requirements. These limitations create significant barriers for pharmaceutical manufacturers seeking consistent high-purity intermediates, particularly when scaling complex molecular architectures where impurities can derail clinical development timelines. The inherent instability of many trifluoromethylation reagents also introduces supply chain vulnerabilities, as specialized handling and storage protocols increase both cost and lead time for critical drug intermediates.
The Novel Approach
The patented methodology (CN111423381A) employs a carefully optimized palladium-catalyzed cascade reaction that eliminates these traditional pain points through strategic reagent selection and mild reaction parameters. 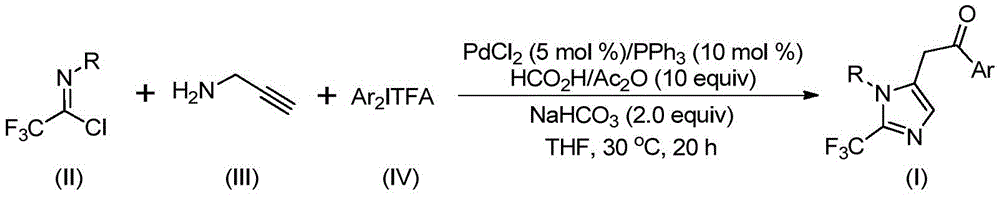 By utilizing trifluoroethylimidoyl chloride as a stable trifluoromethyl synthon and diaryliodonium salts as arylating agents, the process achieves exceptional substrate compatibility across diverse functional groups including halogens, alkyl chains, and nitro substituents. The reaction proceeds through a well-defined mechanism involving base-promoted carbon-nitrogen bond formation, palladium-mediated alkyne amination, and carbonylative cyclization—each step designed to minimize side reactions while maintaining high atom economy. This approach enables the synthesis of previously inaccessible imidazole derivatives with precise regiocontrol at the 1,5 positions, as demonstrated by the five representative compounds shown in the patent examples.
By utilizing trifluoroethylimidoyl chloride as a stable trifluoromethyl synthon and diaryliodonium salts as arylating agents, the process achieves exceptional substrate compatibility across diverse functional groups including halogens, alkyl chains, and nitro substituents. The reaction proceeds through a well-defined mechanism involving base-promoted carbon-nitrogen bond formation, palladium-mediated alkyne amination, and carbonylative cyclization—each step designed to minimize side reactions while maintaining high atom economy. This approach enables the synthesis of previously inaccessible imidazole derivatives with precise regiocontrol at the 1,5 positions, as demonstrated by the five representative compounds shown in the patent examples.
Molecular Mechanism and Purity Control for R&D Excellence
The reaction pathway begins with base-promoted intermolecular carbon-nitrogen bond formation between trifluoroethylimidoyl chloride and propargylamine to generate a trifluoroacetamidine intermediate, followed by isomerization to form an alkenyl palladium species. 
 Subsequent palladium-catalyzed alkyne amination creates an alkyl palladium intermediate that undergoes carbonylation using in situ generated carbon monoxide from formic acid/acetic anhydride mixture. The final oxidative addition with diaryliodonium salt and reductive elimination yields the target imidazole with exceptional regioselectivity.
Subsequent palladium-catalyzed alkyne amination creates an alkyl palladium intermediate that undergoes carbonylation using in situ generated carbon monoxide from formic acid/acetic anhydride mixture. The final oxidative addition with diaryliodonium salt and reductive elimination yields the target imidazole with exceptional regioselectivity.  This cascade mechanism inherently minimizes impurity formation through controlled stepwise transformations, eliminating the need for harsh oxidants or high temperatures that typically generate degradants in conventional syntheses.
This cascade mechanism inherently minimizes impurity formation through controlled stepwise transformations, eliminating the need for harsh oxidants or high temperatures that typically generate degradants in conventional syntheses.
Impurity control is further enhanced by the reaction's tolerance for diverse substituents on both the aryl groups (R and Ar), which can include methyl, tert-butyl, chloro, bromo, trifluoromethyl, or nitro moieties at ortho, meta, or para positions.  The patent demonstrates consistent high-purity outcomes across multiple derivatives (I-1 to I-5), with HRMS data confirming molecular weights within acceptable error margins (e.g., C₁₉H₁₅F₃N₂O calculated 345.1209 vs found 345.1227). The mild reaction conditions (30°C) prevent thermal degradation pathways common in traditional syntheses, while the use of common solvents like THF facilitates straightforward purification via standard column chromatography without specialized equipment. This inherent process robustness ensures consistent impurity profiles across batches—critical for pharmaceutical development where regulatory agencies require stringent control over genotoxic impurities.
The patent demonstrates consistent high-purity outcomes across multiple derivatives (I-1 to I-5), with HRMS data confirming molecular weights within acceptable error margins (e.g., C₁₉H₁₅F₃N₂O calculated 345.1209 vs found 345.1227). The mild reaction conditions (30°C) prevent thermal degradation pathways common in traditional syntheses, while the use of common solvents like THF facilitates straightforward purification via standard column chromatography without specialized equipment. This inherent process robustness ensures consistent impurity profiles across batches—critical for pharmaceutical development where regulatory agencies require stringent control over genotoxic impurities.
Commercial Advantages for Procurement and Supply Chain
This innovative synthesis methodology directly addresses three critical pain points in pharmaceutical manufacturing supply chains: cost inefficiencies from complex purification requirements, extended lead times due to unstable reagents, and scalability limitations of conventional routes. By eliminating transition metal residues through optimized catalyst loading (PdCl₂ at 5 mol%) and leveraging stable, commercially available starting materials, the process reduces both raw material costs and downstream processing expenses while maintaining the high purity standards required for API intermediates.
- Cost reduction in chemical manufacturing: The elimination of expensive transition metal catalysts and hazardous reagents significantly lowers raw material costs while reducing purification complexity. Since the process uses readily available palladium chloride (cheaper than alternative catalysts) and avoids cryogenic conditions, it minimizes energy consumption during manufacturing. The simplified workup procedure—requiring only filtration and standard column chromatography—eliminates costly metal scavenging steps typically needed in conventional syntheses, directly translating to lower production costs per kilogram without compromising quality standards required for pharmaceutical intermediates.
- Reducing lead time for high-purity chemicals: The use of stable, shelf-stable reagents like trifluoroethylimidoyl chloride (synthesized from common aromatic amines) and diaryliodonium salts (prepared from arylphenylboronic acids) eliminates supply chain bottlenecks associated with unstable intermediates. Unlike traditional methods requiring specialized handling of moisture-sensitive reagents, this process utilizes materials with extended shelf lives that can be stored under standard warehouse conditions. The consistent reaction performance across diverse substrates also reduces development timelines for new analogs, enabling faster response to changing project requirements while maintaining reliable delivery schedules for high-purity intermediates.
- Commercial scale-up of complex intermediates: The demonstrated scalability from milligram to gram quantities in the patent examples provides a clear pathway for industrial implementation without fundamental process changes. The mild reaction conditions (30°C) eliminate the need for specialized high-pressure or cryogenic equipment required by alternative methods, significantly reducing capital expenditure for manufacturing facilities. The broad functional group tolerance ensures consistent performance across diverse molecular architectures, while the straightforward purification protocol maintains product quality during scale-up—addressing the critical challenge of preserving purity when transitioning from lab to plant scale for complex heterocyclic intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable API Intermediate Supplier
While the advanced methodology detailed in patent CN111423381A highlights immense potential, executing the commercial scale-up of such complex catalytic pathways requires a proven CDMO partner. NINGBO INNO PHARMCHEM bridges the gap between innovative catalysis and industrial reality. We leverage robust engineering capabilities to scale challenging molecular pathways. Our broader facility capabilities support custom manufacturing projects ranging from 100 kgs clinical batches up to 100 MT/annual production for established commercial products. Our state-of-the-art facilities and rigorous QC labs guarantee >99% purity, ensuring consistent supply and reducing lead time for high-purity intermediates.
Are you evaluating new synthetic routes for your pipeline? Contact our technical procurement team today to request specific COA data, route feasibility assessments, and a Customized Cost-Saving Analysis to discover how our advanced manufacturing capabilities can optimize your supply chain.