Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial API Production
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial API Production
The pharmaceutical industry continuously seeks robust and scalable methodologies for constructing nitrogen-containing heterocycles, particularly those bearing trifluoromethyl groups which enhance metabolic stability and lipophilicity. Patent CN111423381A introduces a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds that addresses many limitations of prior art. This technology leverages a transition metal palladium-catalyzed carbonylation cascade reaction, utilizing readily available starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salts. For R&D directors and procurement managers seeking a reliable pharmaceutical intermediate supplier, this process represents a significant advancement in synthetic efficiency. The reaction operates under remarkably mild conditions, typically at 30°C, avoiding the extreme temperatures or hazardous reagents often associated with trifluoromethylation strategies. By integrating this methodology into your supply chain, organizations can achieve substantial improvements in the cost reduction in API manufacturing while maintaining high purity standards required for drug development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nitrogen-containing heterocycles with trifluoromethyl functional groups has relied heavily on synthons like trifluorodiazoethane or direct reaction with trifluoroethylimide acid halides under restrictive conditions. Conventional literature often cites methods that require hazardous diazo compounds, which pose significant safety risks due to their explosive nature and instability during storage and handling. Furthermore, many existing protocols demand harsh reaction environments, including high temperatures or strong bases, which can lead to poor functional group tolerance and the formation of complex impurity profiles. These factors severely limit the scalability of such processes, making them unsuitable for the commercial scale-up of complex pharmaceutical intermediates. The reliance on specialized, expensive reagents also drives up the cost of goods, creating bottlenecks for procurement teams aiming for cost-effective production routes. Consequently, there has been a persistent need for a safer, more versatile synthetic pathway that can accommodate diverse substrates without compromising yield or safety.
The Novel Approach
The methodology disclosed in the patent offers a transformative solution by employing a palladium-catalyzed carbonylation series reaction that efficiently constructs the imidazole core. This novel approach utilizes cheap and readily available trifluoroethylimide chloride and propargylamine as primary building blocks, reacting them with diaryliodonium salts in the presence of a palladium catalyst. A key innovation is the use of a formic acid and acetic anhydride mixture as a safe, in-situ source of carbon monoxide, eliminating the need for handling toxic CO gas cylinders. The reaction proceeds smoothly in organic solvents like tetrahydrofuran (THF) at a mild temperature of 30°C for approximately 16 to 24 hours. This gentle protocol ensures excellent substrate compatibility, allowing for the introduction of various substituents such as alkyl, halogen, and nitro groups. 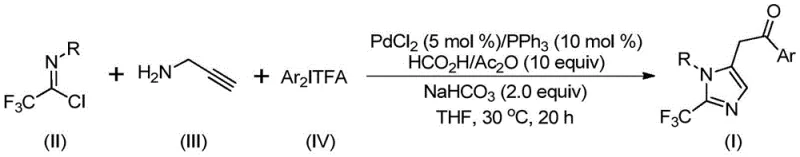
By streamlining the synthetic steps and utilizing stable precursors, this method broadens the utility of trifluoroethylimide acid halides, which previously had limited application potential. The ability to synthesize diversely substituted imidazole compounds through simple substrate design makes this route highly attractive for generating compound libraries in early-stage drug discovery. Moreover, the post-processing is straightforward, involving filtration and standard column chromatography, which facilitates rapid isolation of the target molecules. This operational simplicity, combined with high reaction efficiency, positions this technology as a superior alternative for the production of high-purity pharmaceutical intermediates.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic pathway is crucial for R&D teams optimizing this process for specific analogs. The reaction likely initiates with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and propargylamine, yielding a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization to align for the palladium catalytic cycle. The palladium catalyst, typically PdCl2 paired with triphenylphosphine ligands, facilitates the aminopalladation of the alkyne moiety, generating a vinyl-palladium intermediate. This intermediate then isomerizes to an alkyl-palladium species, setting the stage for the critical carbonylation step. The carbon monoxide, released in situ from the decomposition of the formic acid and acetic anhydride mixture, inserts into the palladium-carbon bond to form an acyl-palladium intermediate. This step is pivotal as it introduces the carbonyl functionality required for the final ring closure.
Following carbonylation, the diaryliodonium salt participates in an oxidative addition step with the acyl-palladium species, forming a high-valent tetravalent palladium intermediate. This oxidative addition is facilitated by the hypervalent iodine species, which acts as an efficient arylating agent under these mild conditions. The catalytic cycle concludes with a reductive elimination step, which releases the final 2-trifluoromethyl substituted imidazole product and regenerates the active palladium catalyst. This intricate cascade allows for the construction of the five-membered heterocyclic ring with precise regiocontrol. 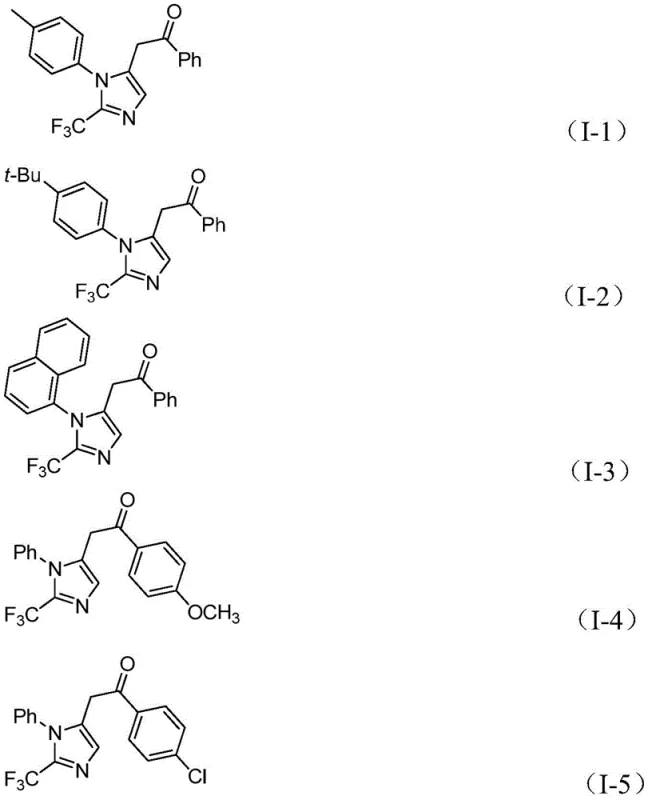
Regarding impurity control, the mild reaction temperature of 30°C significantly suppresses side reactions such as polymerization of the alkyne or decomposition of sensitive functional groups. The use of sodium bicarbonate as a mild base further ensures that acid-sensitive moieties remain intact throughout the transformation. The high selectivity of the palladium catalyst towards the desired cascade pathway minimizes the formation of by-products, simplifying the downstream purification process. For quality control laboratories, this means a cleaner crude reaction profile and higher overall yields, often exceeding 90% for optimized substrates as demonstrated in the patent examples. This level of control is essential for meeting the stringent purity specifications required for clinical grade materials.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
To implement this synthesis effectively, operators should adhere to the specific molar ratios and conditions outlined in the patent data to ensure reproducibility and optimal yield. The process begins by charging a reaction vessel with the palladium catalyst system, specifically palladium chloride and triphenylphosphine, along with sodium bicarbonate and the CO source mixture. The reactants, including the trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salt, are then introduced into the organic solvent, preferably THF, under an inert atmosphere. Maintaining the temperature at 30°C is critical; while the reaction can tolerate a range of 16 to 24 hours, extending beyond this window may increase costs without significant gains in conversion. Detailed standardized synthesis steps follow below for technical reference.
- Mix palladium chloride, triphenylphosphine, sodium bicarbonate, and a formic acid/acetic anhydride mixture in an organic solvent like THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryliodonium salt to the reaction vessel under stirring.
- Maintain the reaction at 30°C for 16 to 24 hours, then filter and purify via column chromatography to isolate the product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers distinct strategic advantages regarding cost structure and supply reliability. The primary driver for cost reduction in pharmaceutical intermediate manufacturing lies in the selection of raw materials and reaction conditions. This process utilizes palladium chloride, which is significantly more economical than many specialized palladium complexes often employed in cross-coupling reactions. Furthermore, the starting materials such as aromatic amines and propargylamine are commodity chemicals widely available in the global market, reducing the risk of supply chain disruptions. The ability to run the reaction at 30°C drastically lowers energy consumption compared to processes requiring reflux or cryogenic conditions, contributing to a lower carbon footprint and reduced utility costs. These factors collectively enable a more competitive pricing model for the final API intermediate.
- Cost Reduction in Manufacturing: The elimination of hazardous reagents like trifluorodiazoethane removes the need for specialized containment equipment and safety protocols, leading to substantial capital expenditure savings. Additionally, the use of in-situ generated carbon monoxide avoids the logistical costs and safety hazards associated with purchasing and storing high-pressure CO gas cylinders. The high atom economy of the cascade reaction ensures that a significant proportion of the starting mass is incorporated into the final product, minimizing waste disposal costs. By simplifying the workup procedure to filtration and chromatography, labor hours and solvent usage are optimized, further driving down the cost per kilogram of the produced material.
- Enhanced Supply Chain Reliability: Since the key building blocks are commercially available and do not require custom synthesis, lead times for raw material acquisition are significantly shortened. The robustness of the reaction against varying substitution patterns on the aryl rings means that a single platform technology can be used to produce a wide array of derivatives without re-validating entirely new processes. This flexibility allows manufacturers to respond quickly to changing demands in drug development pipelines. The scalability of the method from gram to multi-kilogram levels ensures that supply can be ramped up seamlessly as a drug candidate progresses from preclinical to clinical stages, guaranteeing continuity of supply for critical projects.
- Scalability and Environmental Compliance: The mild reaction conditions and the use of common organic solvents like THF facilitate easy adaptation to large-scale reactors without requiring exotic metallurgy or extreme pressure ratings. The waste stream generated is relatively benign compared to processes involving heavy metals or toxic gases, simplifying environmental compliance and wastewater treatment. The high yields reported in the patent examples indicate efficient resource utilization, aligning with green chemistry principles. This environmental compatibility is increasingly important for multinational corporations aiming to meet sustainability goals while maintaining efficient production schedules for complex heterocycles.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation. Understanding these details helps stakeholders assess the feasibility of integrating this route into their existing manufacturing capabilities. The responses cover aspects ranging from reaction optimization to raw material sourcing, providing a comprehensive overview for decision-makers.
Q: What are the key advantages of this palladium-catalyzed method over traditional trifluoromethylation?
A: Unlike traditional methods that often rely on unstable trifluorodiazoethane or harsh conditions, this patent describes a mild process operating at 30°C using stable trifluoroethylimidoyl chlorides and in-situ generated carbon monoxide, significantly enhancing safety and operational simplicity.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method is scalable beyond gram-level quantities. The use of commercially available catalysts like PdCl2 and common solvents like THF, combined with mild temperature requirements, supports feasible commercial scale-up for API manufacturing.
Q: What is the substrate scope for the aryl groups in this reaction?
A: The method demonstrates excellent functional group tolerance, accommodating various substituents on both the imidoyl chloride and the diaryliodonium salt, including methyl, tert-butyl, halogens (chloro, bromo), trifluoromethyl, and nitro groups at ortho, meta, or para positions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug discovery and development. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory results translate into reliable industrial supply. We are equipped with rigorous QC labs and state-of-the-art analytical instrumentation to guarantee stringent purity specifications for every batch of 2-trifluoromethyl imidazole derivatives we produce. Our commitment to quality and consistency makes us a trusted partner for global pharmaceutical companies seeking to secure their supply chains for key intermediates.
We invite you to collaborate with us to leverage this innovative palladium-catalyzed technology for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. Please contact us to request specific COA data and route feasibility assessments for your target molecules. By partnering with us, you gain access to a robust supply network and deep technical expertise dedicated to supporting your journey from bench to bedside.