Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial API Manufacturing
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial API Manufacturing
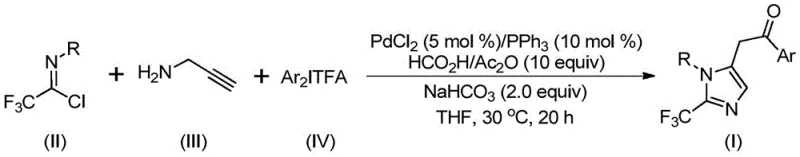
The pharmaceutical industry continuously seeks robust synthetic methodologies to access nitrogen-containing heterocycles functionalized with trifluoromethyl groups, driven by the profound impact of fluorine atoms on the pharmacokinetic profiles of drug candidates. Patent CN111423381B discloses a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds, utilizing a transition metal palladium-catalyzed carbonylation series reaction. This innovation addresses critical challenges in modern medicinal chemistry by enabling the efficient construction of complex imidazole scaffolds under remarkably mild conditions. The introduction of the trifluoromethyl moiety significantly enhances electronegativity, bioavailability, metabolic stability, and lipophilicity, making these intermediates highly valuable for the development of next-generation therapeutics. By leveraging cheap and easily obtainable starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts, this technology offers a streamlined pathway that bridges the gap between academic discovery and commercial manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nitrogen-containing heterocycles bearing trifluoromethyl functional groups has relied heavily on the direct reaction of synthons containing the trifluoromethyl group with suitable substrates, a strategy often fraught with significant operational hurdles. Traditional approaches frequently necessitate the use of specialized and potentially hazardous reagents such as trifluorodiazoethane, which poses safety risks due to its explosive nature and requires stringent handling protocols. Furthermore, many existing methods suffer from limited substrate compatibility, often failing to tolerate diverse functional groups present in complex drug-like molecules, thereby restricting their utility in late-stage functionalization. The requirement for harsh reaction conditions, including extreme temperatures or strong bases, can lead to decomposition of sensitive intermediates and generate difficult-to-remove impurities, complicating the purification process and reducing overall yield. These limitations collectively increase the cost of goods sold and extend the timeline for process development, creating bottlenecks for reliable pharmaceutical intermediate suppliers aiming to deliver high-purity materials efficiently.
The Novel Approach
In stark contrast to conventional techniques, the novel approach detailed in the patent utilizes a sophisticated palladium-catalyzed multicomponent coupling strategy that operates under exceptionally mild conditions, typically at 30°C for 16 to 24 hours. This method ingeniously employs trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts as the core building blocks, facilitated by a catalytic system comprising palladium chloride and triphenylphosphine. The reaction design allows for the in situ generation of carbon monoxide from formic acid and acetic anhydride, effectively serving as a safe and controllable carbonyl source without the need for high-pressure gas cylinders. This strategic integration of reagents not only simplifies the operational setup but also dramatically improves the safety profile of the synthesis. The versatility of this approach is evidenced by its ability to accommodate a wide range of substituents on the aryl rings, including alkyl, halogen, trifluoromethyl, and nitro groups, ensuring broad applicability across diverse chemical spaces. Consequently, this methodology represents a significant leap forward in cost reduction in pharmaceutical intermediate manufacturing by maximizing atom economy and minimizing waste generation.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The mechanistic pathway of this transformation involves a complex yet elegant sequence of organometallic steps orchestrated by the palladium catalyst, beginning with the formation of a key carbon-nitrogen bond promoted by the alkaline additive. Initially, an intermolecular reaction between the amine and the imidoyl chloride likely generates a trifluoroacetamidine intermediate, which subsequently undergoes isomerization to align the reactive centers for cyclization. The palladium catalyst then engages with the alkyne moiety of the propargylamine derivative through a palladation event, forming a vinyl-palladium intermediate that sets the stage for ring closure. This intermediate further isomerizes to an alkyl-palladium species, positioning the metal center for the crucial carbonylation step where carbon monoxide, released from the formic acid/acetic anhydride mixture, inserts into the palladium-carbon bond. The resulting acyl-palladium intermediate then undergoes oxidative addition with the diaryl iodonium salt, generating a high-valent tetravalent palladium species that is pivotal for the final bond formation. The catalytic cycle concludes with a reductive elimination step that releases the desired 2-trifluoromethyl-substituted imidazole product and regenerates the active palladium catalyst for subsequent turnover.
From an impurity control perspective, the mild reaction temperature of 30°C plays a vital role in suppressing side reactions that typically plague high-temperature processes, such as polymerization of the alkyne or decomposition of the diazo-like intermediates. The use of sodium bicarbonate as a base ensures a buffered environment that facilitates the necessary deprotonation steps without promoting excessive hydrolysis of the sensitive imidoyl chloride or the final imidazole product. Furthermore, the choice of tetrahydrofuran (THF) as the preferred organic solvent provides an optimal balance of solubility for all reactants and the catalyst system, ensuring homogeneous reaction conditions that minimize the formation of insoluble byproducts. The specificity of the oxidative addition step with the diaryl iodonium salt also contributes to high regioselectivity, ensuring that the aryl group is installed at the correct position on the imidazole ring. This precise control over the reaction trajectory results in a clean crude reaction profile, significantly reducing the burden on downstream purification units and enhancing the overall purity of the high-purity pharmaceutical intermediates produced.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The practical execution of this synthesis is designed for reproducibility and ease of scale-up, making it an ideal candidate for contract development and manufacturing organizations. The protocol involves charging a reaction vessel with the palladium catalyst, ligand, and additives, followed by the sequential addition of the three main coupling partners in an appropriate organic solvent. The reaction proceeds spontaneously at near-ambient temperature, eliminating the need for energy-intensive heating or cooling systems, which is a significant advantage for green chemistry initiatives. Upon completion, the workup procedure is straightforward, involving simple filtration to remove inorganic salts and catalyst residues, followed by standard silica gel column chromatography to isolate the pure product. For detailed operational parameters and specific stoichiometric ratios optimized for different substrates, please refer to the standardized synthesis guide below.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, and formic acid in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 16 to 24 hours, followed by filtration and column chromatography purification to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers substantial strategic benefits that extend beyond mere technical feasibility. The reliance on commodity chemicals such as propargylamine and commercially available palladium salts drastically reduces the dependency on exotic or custom-synthesized reagents, thereby stabilizing the supply chain against market volatility. The elimination of hazardous reagents like trifluorodiazoethane not only lowers regulatory compliance costs but also simplifies logistics and storage requirements, allowing for more flexible inventory management. Moreover, the high reaction efficiency and broad substrate tolerance mean that a single manufacturing platform can be utilized to produce a diverse library of analogues, optimizing asset utilization and reducing the capital expenditure required for dedicated production lines. These factors collectively contribute to a more resilient and cost-effective supply network for critical drug substances.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of inexpensive catalysts like palladium chloride and triphenylphosphine, which are significantly cheaper than many specialized noble metal complexes used in alternative cross-coupling reactions. The ability to run the reaction at 30°C translates to massive energy savings compared to processes requiring reflux or cryogenic conditions, directly impacting the utility costs associated with large-scale production. Additionally, the high yields reported across various substrates minimize the loss of valuable starting materials, ensuring that the cost per kilogram of the final API intermediate remains competitive. By streamlining the synthetic route to fewer steps and avoiding protective group manipulations, the overall material throughput is maximized, delivering substantial cost savings in raw material procurement.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures consistent quality and availability, which is paramount for maintaining uninterrupted drug supply chains. Since the key starting materials are widely available from multiple global vendors, the risk of single-source bottlenecks is effectively mitigated, providing procurement teams with greater negotiating power and flexibility. The simplicity of the post-treatment process, which avoids complex extractions or distillations, reduces the turnaround time for batch release, allowing for faster response to market demand fluctuations. This reliability makes the technology an attractive option for securing long-term contracts for reliable pharmaceutical intermediate supplier partnerships, ensuring that clinical and commercial timelines are met without delay.
- Scalability and Environmental Compliance: Scaling this process from gram to multi-kilogram quantities is facilitated by the homogeneous nature of the reaction and the absence of gas handling issues, as the carbon monoxide is generated in situ from liquid precursors. This inherent safety feature simplifies the engineering controls required for commercial scale-up of complex pharmaceutical intermediates, reducing the time and cost associated with technology transfer. From an environmental standpoint, the use of formic acid as a CO source and the generation of benign byproducts align with green chemistry principles, helping manufacturers meet increasingly stringent environmental regulations. The reduced solvent usage and simplified waste streams further lower the environmental footprint, supporting corporate sustainability goals while maintaining operational efficiency.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis method, derived directly from the experimental data and scope defined in the patent literature. Understanding these nuances is essential for R&D teams planning to integrate this technology into their existing workflows or for quality assurance personnel evaluating the process robustness. The answers provided reflect the specific conditions and outcomes observed during the development of this methodology, ensuring accuracy and relevance for technical decision-making.
Q: What are the key advantages of this palladium-catalyzed method over traditional trifluoromethylation?
A: This method operates under mild conditions (30°C) using readily available starting materials like propargylamine and trifluoroethylimidoyl chloride, avoiding the harsh conditions and specialized reagents often required in direct trifluoromethylation protocols.
Q: What is the substrate scope for the aryl groups in this synthesis?
A: The process demonstrates excellent compatibility with various substituted aryl groups, including those with electron-donating groups like methyl and methoxy, as well as electron-withdrawing groups such as halogens, nitro, and trifluoromethyl substituents.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method is scalable to gram levels and potentially industrial scales due to the use of cheap catalysts (PdCl2/PPh3), simple workup procedures involving filtration and chromatography, and high reaction efficiency.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed carbonylation technology in accelerating the development of fluorinated drug candidates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from laboratory benchtop to full-scale manufacturing. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl imidazole intermediate delivered meets the highest industry standards. We are committed to leveraging our technical expertise to optimize this route further, tailoring the process to your specific volume and quality requirements while maintaining full regulatory compliance.
We invite you to collaborate with us to unlock the full commercial potential of your fluorinated imidazole programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis that details how implementing this specific synthetic route can optimize your budget without compromising on quality. Please contact us today to request specific COA data for our reference standards and to discuss route feasibility assessments for your target molecules, ensuring a seamless path to market for your innovative therapies.