Advanced Synthesis of Florfenicol Intermediates: A Scalable Route for Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust synthetic pathways that balance high yield with operational simplicity, particularly for critical antibiotic precursors. Patent CN110330463B introduces a transformative preparation method for a key florfenicol intermediate, specifically targeting the synthesis of fluoride oxazoline derivatives. This innovation departs from legacy methodologies by replacing the cumbersome Ishikawa reagent system with a streamlined two-step sequence involving thionyl chloride chlorination followed by potassium fluoride substitution. For R&D directors and process chemists, this represents a significant leap forward, offering a route that achieves mass yields exceeding 96.0% while drastically simplifying the reaction infrastructure. The technical breakthrough lies not just in the reagents chosen, but in the specific activation of the fluorinating agent, which overcomes the kinetic barriers typically associated with nucleophilic fluorination in organic synthesis. By adopting this approach, manufacturers can secure a more reliable florfenicol intermediate supplier pipeline that is less susceptible to the supply chain volatility of exotic fluorinating agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of florfenicol intermediates has relied heavily on the methodology described in patents such as US5382673, which utilizes an Ishikawa reagent for the critical fluorination step. This conventional approach is fraught with logistical and chemical inefficiencies that burden large-scale manufacturing operations. The process necessitates the in-situ generation of the fluorinating agent by slowly introducing hexafluoropropylene into a mixture of dichloromethane and diethylamine at temperatures below 10°C, followed by an extensive stirring period of up to 18 hours at room temperature. Subsequently, the cyclic oxazoline substrate must react with this freshly prepared reagent at elevated temperatures around 100°C for several hours. This multi-stage protocol not only demands sophisticated industrial equipment capable of handling hazardous gases and extreme thermal conditions but also results in the formation of stubborn amide by-products. These amide impurities complicate downstream purification, often requiring rigorous washing with alkaline solutions and multiple crystallization steps, which inevitably erodes the overall process yield and increases the environmental footprint of the manufacturing facility.
The Novel Approach
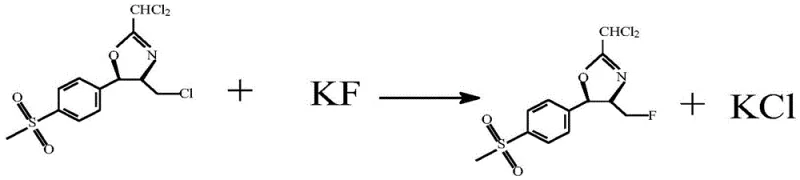
In stark contrast, the novel approach detailed in the provided patent data revolutionizes this synthesis by decoupling the activation and substitution steps into a highly efficient sequence. Instead of generating a complex fluorinating reagent, the process first converts the hydroxyl group of the cyclic oxazoline into a superior leaving group via chlorination using thionyl chloride. This transformation is rapid and clean, producing gaseous by-products like sulfur dioxide and hydrogen chloride that are easily vented or scrubbed, leaving behind a reactive chloro-intermediate. The subsequent fluorination step utilizes potassium fluoride, a commodity chemical, rather than a custom-synthesized reagent. Crucially, the patent specifies a pre-treatment of the potassium fluoride involving reflux in methanol and vacuum drying, which creates a fluffy, high-surface-area solid. This "specialized" potassium fluoride exhibits dramatically enhanced nucleophilicity, allowing the substitution reaction to proceed to completion within one hour at a mild 30°C. This shift eliminates the need for high-temperature refluxes and complex reagent preparation, directly addressing the pain points of cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Thionyl Chloride Activation and KF Substitution
The mechanistic elegance of this route begins with the activation of the primary alcohol on the oxazoline ring. When thionyl chloride is introduced to the dichloromethane solution of the cyclic compound at a controlled temperature of 15°C, it reacts with the hydroxyl group to form a chlorosulfite intermediate. This intermediate rapidly collapses, releasing sulfur dioxide gas and displacing the oxygen with a chlorine atom via an SN2-like mechanism. The precision of temperature control here is paramount; maintaining the reaction between 10°C and 20°C prevents the degradation of the sensitive oxazoline ring while ensuring complete conversion of the alcohol. Experimental data indicates that a molar ratio of cyclic compound to thionyl chloride of 1:1.4 is optimal, providing a slight excess of the chlorinating agent to drive the equilibrium forward without generating excessive acidic waste. The result is a chloromethyl-oxazoline intermediate that is primed for nucleophilic attack, possessing a leaving group that is far more labile than the original hydroxyl moiety.
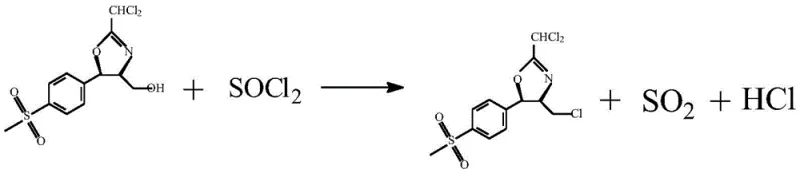
Following chlorination, the introduction of the specialized potassium fluoride initiates the critical halogen exchange. Standard potassium fluoride often suffers from poor solubility in organic solvents and strong lattice energy, rendering it unreactive. However, the patented pre-treatment—refluxing in methanol followed by vacuum drying at 100°C—disrupts the crystal lattice and removes hydration water, creating a highly active, anhydrous fluoride source. When this activated salt encounters the chloromethyl intermediate in dichloromethane, the fluoride ion acts as a potent nucleophile, displacing the chlorine atom to form the carbon-fluorine bond. This step is remarkably tolerant, proceeding efficiently at 30°C with a molar ratio of 1:1.2 (intermediate to KF). The mechanism avoids the formation of the amide impurities seen in the Ishikawa route because no amine bases are present to compete with the fluoride or react with the carbonyl equivalents. The outcome is a clean conversion to the fluoride oxazoline, preserving the stereochemical integrity of the chiral centers essential for the biological activity of the final antibiotic.
How to Synthesize Fluoride Oxazoline Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters identified in the patent examples to ensure reproducibility and maximum yield. The process is divided into three distinct operational phases: the preparation of the activated fluorinating agent, the chlorination of the substrate, and the final substitution reaction. Operators must pay particular attention to the physical state of the potassium fluoride, as the "fluffy flocculent" texture achieved through the methanol reflux and drying cycle is a critical quality attribute that dictates reaction kinetics. Furthermore, the recovery of dichloromethane should be managed carefully, initially at normal pressure and subsequently under reduced pressure at elevated temperatures (60-70°C) to ensure complete solvent removal without degrading the thermally sensitive product. The detailed standardized synthesis steps below outline the precise stoichiometry and thermal profiles required for successful commercial scale-up of complex pharmaceutical intermediates.
- Dissolve cyclic oxazoline in dichloromethane (1: 15-25 ratio) and add thionyl chloride (1:1.3-1.5 ratio) at 10-20°C to form the chlorinated intermediate.
- Prepare specialized potassium fluoride by refluxing in methanol, recovering solvent under vacuum, and drying at 100°C to increase surface area and reactivity.
- React the chlorinated intermediate with the specialized potassium fluoride (1: 1.1-1.3 ratio) at 10-50°C for 0.5-1.5 hours to achieve fluorination.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers compelling strategic advantages that extend beyond simple yield metrics. The most immediate impact is the drastic simplification of the raw material portfolio. By eliminating the need for hexafluoropropylene and diethylamine for reagent synthesis, facilities can reduce their dependency on specialized gas suppliers and minimize the inventory of hazardous volatile organic compounds. This simplification directly correlates to enhanced supply chain reliability, as potassium fluoride and thionyl chloride are widely available commodity chemicals with stable global supply networks. Furthermore, the reduction in process steps—from a multi-day sequence involving reagent generation to a streamlined two-step one-pot style workflow—significantly shortens the manufacturing cycle time. This agility allows producers to respond more rapidly to market demand fluctuations for veterinary antibiotics, ensuring consistent availability of high-purity florfenicol intermediates without the bottlenecks associated with complex reagent preparation.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the elimination of the expensive and labor-intensive Ishikawa reagent preparation. In the traditional route, significant resources are consumed in cooling, stirring, and handling hexafluoropropylene for nearly a day before the actual fluorination even begins. By replacing this with a direct substitution using activated potassium fluoride, the process removes an entire unit operation, thereby reducing energy consumption, labor hours, and reactor occupancy time. Additionally, the avoidance of amide by-products means that downstream purification is less resource-intensive, requiring fewer wash cycles and less solvent for recrystallization. While specific percentage savings depend on local utility costs, the qualitative reduction in raw material investment and processing time translates to substantial cost savings per kilogram of finished intermediate.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the robustness of the reaction conditions. The new method operates at mild temperatures (15°C for chlorination, 30°C for fluorination), reducing the thermal stress on equipment and minimizing the risk of runaway reactions or thermal degradation that can lead to batch failures. The use of solid potassium fluoride, which can be prepared in bulk and stored, decouples the fluorination step from the immediate availability of gaseous reagents. This inventory flexibility allows manufacturers to maintain buffer stocks of critical reagents, mitigating the risk of production stoppages due to logistics delays. Consequently, partners can rely on a more predictable production schedule, ensuring that the flow of high-purity pharmaceutical intermediates remains uninterrupted even during periods of global supply constraint.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this route is inherently greener and easier to scale. The by-products of the chlorination step are sulfur dioxide and hydrogen chloride, both of which are acidic gases that can be efficiently captured and neutralized using standard caustic scrubber systems found in most modern chemical plants. This contrasts sharply with the amide waste generated by the Ishikawa method, which often requires complex aqueous workups and generates saline wastewater that is difficult to treat. The simplified waste stream facilitates easier regulatory compliance and reduces the burden on effluent treatment plants. Moreover, the reaction exotherms are manageable, and the use of dichloromethane as a single solvent throughout the process simplifies solvent recovery and recycling loops, making the technology highly suitable for scaling from pilot batches to multi-ton commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and operational inquiries regarding the implementation of this fluorination technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aiming to clarify the practical implications for industrial adoption. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this route into existing manufacturing lines. The answers focus on the critical success factors identified during the optimization studies, such as temperature sensitivity and reagent activation.
Q: Why is this new fluorination method superior to the traditional Ishikawa reagent route?
A: The traditional method requires the complex in-situ preparation of Ishikawa reagent using hexafluoropropylene and diethylamine, which involves long reaction times (up to 18 hours) and generates difficult-to-remove amide by-products. The new method utilizes thionyl chloride and potassium fluoride, simplifying the process, reducing raw material investment, and generating gaseous by-products (SO2, HCl) that are easier to handle industrially.
Q: What are the critical parameters for maximizing yield in this synthesis?
A: Critical parameters include maintaining the chlorination temperature strictly between 10-20°C (optimally 15°C) to prevent side reactions, using a molar ratio of cyclic compound to thionyl chloride of 1:1.4, and employing 'specialized' potassium fluoride prepared via methanol reflux to ensure high surface area and reactivity during the substitution step at 30°C.
Q: How does this process impact the purity profile of the final florfenicol intermediate?
A: By avoiding the formation of amide impurities associated with the Ishikawa reagent route, this novel process significantly reduces the complexity of the impurity spectrum. The use of gaseous by-products allows for easier separation, resulting in a cleaner crude product that requires less intensive refining, thereby supporting higher overall purity specifications suitable for API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Florfenicol Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to advanced synthetic routes requires a partner with deep technical expertise and proven manufacturing capabilities. As a leading CDMO, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel fluorination method are fully realized in a commercial setting. Our facilities are equipped with the necessary corrosion-resistant reactors and gas scrubbing systems to handle thionyl chloride and acidic by-products safely and efficiently. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of florfenicol intermediate meets the exacting standards required for veterinary pharmaceutical applications. Our commitment to quality ensures that the impurity profiles remain minimal, leveraging the inherent cleanliness of the KF substitution mechanism.
We invite procurement leaders and R&D teams to collaborate with us to optimize their supply chains for antibiotic intermediates. By leveraging this patented technology, we can offer a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how the elimination of the Ishikawa reagent impacts your bottom line. We encourage you to contact our technical procurement team to request specific COA data from our recent pilot runs and to discuss route feasibility assessments for your upcoming projects. Together, we can secure a sustainable and cost-effective supply of high-value pharmaceutical building blocks.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →