Revolutionizing Florfenicol Intermediate Production: A Deep Dive into Advanced Catalytic Synthesis and Commercial Scalability
The global demand for broad-spectrum veterinary antibiotics continues to surge, driven by the expanding livestock industry and the critical need for effective disease management in food-producing animals. At the forefront of this market is Florfenicol, a potent chloramphenicol analog that has become a cornerstone in treating respiratory and enteric infections. However, the economic viability of Florfenicol production hinges entirely on the efficiency of its key precursor synthesis. Patent CN101550110B introduces a transformative preparation method for D-threo-2-(dichloromethyl)-4,5-dihydro-5-(p-(methylsulfonyl)phenyl)-4-oxazole methanol, a pivotal intermediate that dictates the stereochemical integrity and ultimate yield of the final antibiotic. This technical insight report analyzes the profound implications of this patented methodology, which shifts the paradigm from multi-step isolation processes to a streamlined, cost-effective continuous workflow. By leveraging specific catalytic conditions and solvent engineering, this route addresses long-standing bottlenecks in chiral intermediate manufacturing, offering a compelling value proposition for R&D directors seeking robust synthetic pathways and procurement managers aiming for sustainable cost structures in the competitive veterinary pharmaceutical landscape.
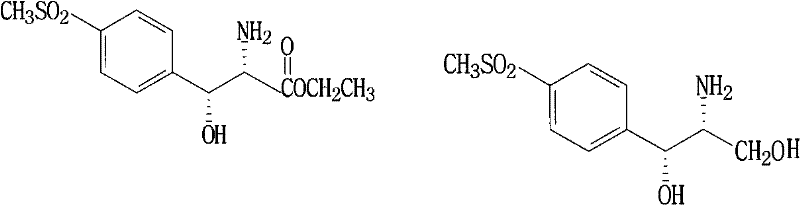
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Florfenicol intermediates has been plagued by operational inefficiencies and excessive material consumption. Traditional routes, such as those referenced in prior art like CN1233244A, typically involve the reduction of D-p-methylsulfinyl phenyl ethyl serinate to an amino-diol intermediate (ADS), followed by a distinct acidification and isolation step. This fragmented approach necessitates multiple unit operations, including filtration, drying, and re-dissolution, each introducing potential yield losses and increasing the cumulative exposure of the chiral center to racemization risks. Furthermore, conventional cyclization strategies often rely on harsh acidic conditions or expensive catalysts to facilitate the formation of the oxazole ring, frequently resulting in a mixture of D-threo and D-erythro isomers that require cumbersome separation protocols. The reliance on aqueous isopropanol systems for isomerization in older methods adds another layer of complexity, requiring precise temperature controls around 80°C and extended reaction times to achieve acceptable conversion ratios. These legacy processes not only inflate the capital expenditure required for reactor occupancy but also generate substantial volumes of solvent waste, creating environmental compliance burdens that modern chemical enterprises strive to minimize.
The Novel Approach
In stark contrast, the methodology outlined in CN101550110B presents a cohesive, integrated strategy that dramatically simplifies the manufacturing topology. The core innovation lies in the seamless transition from reduction to cyclization without isolating the unstable amino-diol intermediate. By employing potassium borohydride in a methanol medium, the process generates a stable borate complex intermediate (Compound V) that protects the sensitive functional groups during the initial transformation. Instead of quenching and isolating this species, the protocol ingeniously recycles a portion of the methanol solvent and introduces glycerol to create a mixed solvent system that supports the subsequent ring-closure reaction. This eliminates the need for intermediate drying and handling, significantly reducing the cycle time and labor intensity associated with batch processing. Moreover, the process leverages the ammonia byproduct generated during the cyclization with dichloroacetonitrile to drive the thermodynamic isomerization of the undesired iso-form (Compound IV) into the target D-threo product (Compound III). This self-sufficient chemical loop removes the necessity for external ammoniation steps, showcasing a level of atom economy and process intensification that sets a new benchmark for veterinary drug intermediate synthesis.
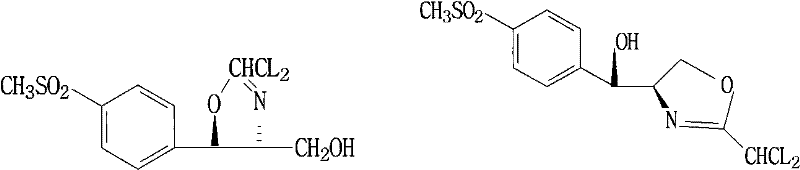
Mechanistic Insights into Potassium Borohydride Reduction and Cyclization
To fully appreciate the technical superiority of this route, one must dissect the intricate chemical mechanisms governing the transformation. The initial reduction phase utilizes potassium borohydride (KBH4) not merely as a hydride source but as a structural stabilizer. Upon reacting with the ester group of the starting material, KBH4 forms a transient organoborate species (Compound V), as depicted in the structural analysis. This coordination is crucial because it prevents the premature degradation or racemization of the chiral center adjacent to the sulfone group, which is highly susceptible to epimerization under basic conditions. The stoichiometry of the reducing agent is tightly controlled, typically ranging from 24 to 27 parts per 100 parts of substrate, ensuring complete conversion while minimizing excess boron residues that could complicate downstream purification. The reaction temperature is maintained between 30°C and 50°C, a moderate thermal window that balances reaction kinetics with the thermal stability of the borate complex, preventing exothermic runaways that could compromise safety and selectivity in large-scale reactors.
The subsequent cyclization and isomerization phases are governed by precise pH control and solvent polarity effects. Once the reduction is complete, the reaction mixture is neutralized to a pH between 7.0 and 10.0, with a preferred optimum of 7.0 to 8.0. This specific pH window is critical; if the environment is too acidic (pH < 7.0), the borate complex decomposes prematurely, exposing the free amine to side reactions. Conversely, if the pH exceeds 10.0, the stability of the intermediate is compromised, leading to a sharp decline in yield. The addition of dichloroacetonitrile in the glycerol-methanol mixed solvent initiates the ring closure, forming the oxazole moiety. Uniquely, this step releases ammonia as a byproduct, which remains dissolved in the polar solvent matrix. This in-situ ammonia acts as a mild base catalyst, facilitating the reversible isomerization of the kinetic product (Compound IV) into the thermodynamically more stable D-threo isomer (Compound III). This mechanistic elegance ensures high diastereoselectivity without the need for external chiral auxiliaries or harsh equilibration conditions, resulting in a final product with specific rotation values consistent with high optical purity requirements for pharmaceutical applications.
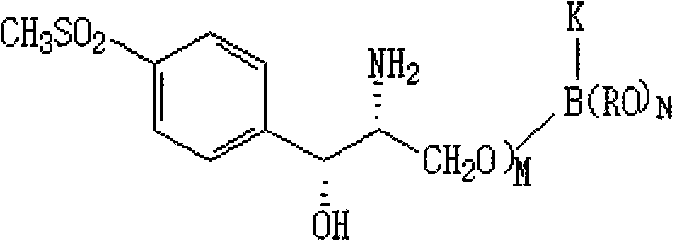
How to Synthesize D-Threo-Oxazole Methanol Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters regarding solvent ratios and pH adjustments to ensure reproducibility at scale. The process begins with the dissolution of the serinate ester in anhydrous methanol, followed by the controlled addition of the reducing agent under inert atmosphere to prevent moisture interference. Following the reduction, the partial removal of methanol and introduction of glycerol creates the necessary viscosity and polarity for the cyclization step. Acid neutralization must be performed slowly with real-time pH monitoring to avoid local over-acidification.
- Dissolve D-p-methylsulfinyl phenyl ethyl serinate in methanol and reduce with potassium borohydride at 30-50°C to form the borate intermediate.
- Recycle methanol and add glycerol, then neutralize the solution to pH 7.0-10.0 using acid to stabilize the intermediate structure.
- Add dichloroacetonitrile to induce cyclization and isomerization, utilizing in-situ generated ammonia to convert the iso-form to the target D-threo product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel synthetic route offers tangible strategic advantages beyond mere technical curiosity. The primary value driver is the significant reduction in operational complexity, which directly translates to lower manufacturing overheads. By eliminating the isolation and drying of the amino-diol intermediate, the process reduces energy consumption associated with vacuum distillation and oven drying, while also minimizing the loss of material typically incurred during solid-liquid separations. The use of glycerol, a widely available and cost-effective green solvent, further enhances the economic profile by replacing more expensive or hazardous organic solvents often required in traditional isomerization steps. Additionally, the simplified workflow shortens the overall production cycle time, allowing for faster turnover of reactor assets and improved responsiveness to fluctuating market demands for veterinary antibiotics. This agility is crucial in an industry where supply continuity can be disrupted by raw material shortages or regulatory inspections.
- Cost Reduction in Manufacturing: The elimination of intermediate isolation steps fundamentally alters the cost structure of the intermediate. By avoiding the filtration, washing, and drying of the unstable amino-diol, manufacturers save on both labor and utility costs. Furthermore, the high atom economy of the in-situ isomerization reduces the requirement for additional reagents and solvents that would otherwise be needed to force equilibrium in a separate vessel. The patent data indicates a substantial decrease in raw material costs per gram of product compared to legacy methods, primarily driven by higher overall yields and reduced solvent make-up rates. This efficiency allows suppliers to offer more competitive pricing models without compromising margin, providing a buffer against volatility in the prices of key starting materials like the serinate ester.
- Enhanced Supply Chain Reliability: Robustness in chemical synthesis is paramount for maintaining uninterrupted supply chains. This method demonstrates superior tolerance to minor variations in reaction conditions due to the stabilizing effect of the borate intermediate, reducing the risk of batch failures that can lead to costly delays. The reliance on commodity chemicals such as methanol, glycerol, and potassium borohydride ensures that raw material sourcing is not bottlenecked by specialty supplier constraints. Moreover, the simplified purification process, which typically involves standard crystallization or solvent wash techniques, facilitates quicker quality control release times. This predictability enables supply chain planners to maintain leaner inventory levels while still meeting just-in-time delivery commitments to downstream API manufacturers.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces heat transfer and mixing challenges that are absent in the laboratory. However, the moderate temperature range (30-50°C) and the use of non-volatile glycerol mitigate many of the safety risks associated with exothermic reactions on a multi-ton scale. The reduction in solvent usage and the avoidance of heavy metal catalysts align with increasingly stringent environmental regulations regarding waste discharge and VOC emissions. The aqueous workup and potential for solvent recycling further enhance the sustainability profile of the manufacturing site. For multinational corporations with strict ESG mandates, adopting this greener synthesis route supports corporate sustainability goals while ensuring long-term regulatory compliance in diverse global jurisdictions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on critical process parameters and quality outcomes. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer or capacity expansion projects.
Q: What is the critical pH range for the cyclization step in this synthesis?
A: The patent data specifies a critical pH range of 7.0 to 10.0, with an optimal window between 7.0 and 8.0. Deviating below 7.0 or above 10.0 compromises the stability of the borate intermediate, leading to significantly reduced yields and difficulty in the subsequent ring-closure reaction.
Q: How does this method improve upon conventional florfenicol intermediate synthesis?
A: Conventional methods often require isolating the amino-diol intermediate (ADS) and performing separate isomerization steps. This novel approach combines reduction and cyclization, utilizing in-situ generated ammonia from the reaction itself to drive isomerization, thereby reducing processing steps, operational complexity, and overall production costs.
Q: What are the purity specifications for the final intermediate produced via this route?
A: According to the technical data, the resulting D-threo-2-(dichloromethyl)-4,5-dihydro-5-(p-(methylsulfonyl)phenyl)-4-oxazole methanol achieves a content of greater than 98%, with moisture levels controlled below 0.1% and a specific rotation of [α]20D +11.5°, ensuring high suitability for downstream antibiotic synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable D-Threo-Oxazole Methanol Supplier
At NINGBO INNO PHARMCHEM, we recognize that the theoretical advantages of a patent must be translated into practical, commercial reality to deliver true value. As a premier CDMO partner, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate pH controls and solvent management required by this synthesis are executed with precision. Our facility is equipped with rigorous QC labs and stringent purity specifications that guarantee every batch of D-threo-2-(dichloromethyl)-4,5-dihydro-5-(p-(methylsulfonyl)phenyl)-4-oxazole methanol meets the exacting standards required for veterinary API synthesis. We understand that consistency is key, and our process engineering team is dedicated to optimizing these reaction conditions to maximize yield and minimize impurity profiles, providing you with a reliable foundation for your final drug product.
We invite you to collaborate with us to leverage this advanced technology for your supply chain. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out for specific COA data and route feasibility assessments to verify how this streamlined synthesis can enhance your operational efficiency. Let us help you secure a competitive edge in the veterinary pharmaceutical market through superior chemistry and dependable supply.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →