Advanced Palladium-Catalyzed Carbonylation for Scalable Quinazolinone Pharmaceutical Intermediates
Advanced Palladium-Catalyzed Carbonylation for Scalable Quinazolinone Pharmaceutical Intermediates
The pharmaceutical industry continuously seeks robust synthetic methodologies to access nitrogen-containing heterocycles, particularly quinazolinones, which serve as privileged scaffolds in medicinal chemistry. As detailed in the recent patent CN113045503A, a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone compounds has been disclosed, offering a transformative approach to constructing these vital pharmacophores. Quinazolinone derivatives are ubiquitous in nature and synthetic drugs, exhibiting a broad spectrum of biological activities ranging from antifungal and antibacterial to antiviral and anticancer properties. The strategic introduction of a trifluoromethyl group into these heterocyclic systems significantly enhances their physicochemical profiles, including metabolic stability, lipophilicity, and bioavailability, making them highly desirable targets for modern drug discovery programs. This new technology addresses the critical need for efficient, scalable, and cost-effective routes to these high-value intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinones has been fraught with significant technical and economic challenges that hinder large-scale manufacturing. Traditional pathways often rely on the cyclization of anthranilamide with ethyl trifluoroacetate, trifluoroacetic anhydride, or trifluoroacetic acid under varying conditions, which frequently necessitate harsh reaction environments and generate substantial waste. Alternative strategies involving the cyclization of anthranilic acid esters with unstable trifluoroacetamides or the use of isatoic anhydride suffer from poor atom economy and the requirement for difficult-to-handle reagents. Furthermore, methods promoted by coupling agents like T3P often involve expensive substrates that require pre-activation steps, leading to increased operational complexity and reduced overall yields. These conventional limitations result in narrow substrate scopes and inconsistent purity profiles, creating bottlenecks for reliable pharmaceutical intermediate suppliers aiming to deliver high-quality materials consistently.
The Novel Approach
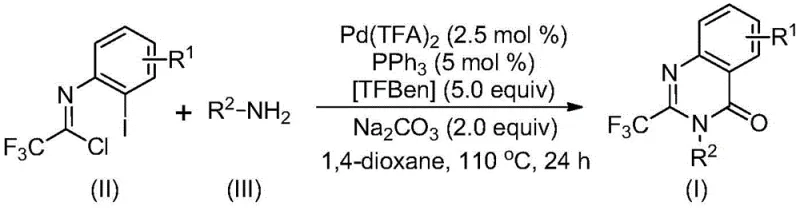
In stark contrast to these legacy methods, the novel approach disclosed in the patent utilizes a transition metal palladium-catalyzed carbonylation tandem reaction that fundamentally simplifies the synthetic architecture. By employing cheap and easily obtainable trifluoroethylimidoyl chloride and various amines as starting materials, this method bypasses the need for pre-activated substrates or hazardous gaseous carbon monoxide. The reaction proceeds efficiently in an organic solvent such as 1,4-dioxane at 110 °C, utilizing TFBen as a solid carbon monoxide substitute to drive the carbonylation process. This innovative strategy not only streamlines the operation but also dramatically expands the substrate compatibility, allowing for the synthesis of diverse substituted trifluoromethyl quinazolinone compounds through rational substrate design. The practicality of this method is significantly widened, offering a versatile platform for generating complex molecular libraries.
Mechanistic Insights into Pd-Catalyzed Carbonylation Tandem Reaction
The mechanistic pathway of this transformation is a sophisticated orchestration of organometallic steps that ensures high efficiency and selectivity. The reaction likely initiates with an alkali-promoted intermolecular carbon-nitrogen bond coupling between the amine and the imidoyl chloride to form a trifluoroacetamidine derivative in situ. Subsequently, the palladium catalyst, generated from palladium trifluoroacetate and triphenylphosphine, undergoes oxidative addition into the carbon-iodine bond of the aromatic ring, forming a key divalent palladium intermediate. Under the thermal conditions provided, the TFBen additive decomposes to release carbon monoxide, which then inserts into the carbon-palladium bond to generate an acyl palladium species. This acyl intermediate is primed for intramolecular nucleophilic attack by the nitrogen base, promoted by the presence of sodium carbonate, leading to the formation of a seven-membered ring palladium intermediate. The cycle concludes with a reductive elimination step that releases the final 2-trifluoromethyl-substituted quinazolinone product and regenerates the active palladium catalyst for the next turnover.
From an impurity control perspective, this mechanism offers distinct advantages over non-catalytic thermal cyclizations. The specificity of the palladium insertion and the controlled release of carbon monoxide from TFBen minimize side reactions such as polymerization or non-selective acylation that often plague traditional methods. The use of a defined ligand system (triphenylphosphine) stabilizes the palladium center, preventing the formation of palladium black and ensuring a homogeneous catalytic environment that favors the desired cyclization pathway. This precise control over the reaction trajectory results in cleaner crude reaction mixtures, thereby reducing the burden on downstream purification processes and ensuring that the final high-purity pharmaceutical intermediates meet stringent quality specifications required by global regulatory bodies.
How to Synthesize 2-Trifluoromethyl Substituted Quinazolinone Efficiently
The execution of this synthesis requires careful attention to reagent stoichiometry and reaction parameters to maximize yield and purity. The protocol involves mixing the palladium catalyst, ligand, base, CO source, and substrates in a sealed vessel to maintain the integrity of the reaction environment. While the general procedure is robust, optimizing the molar ratios of the amine to the imidoyl chloride is crucial for driving the equilibrium towards the product. For a comprehensive understanding of the standardized operating procedures, including specific workup techniques and purification protocols, please refer to the detailed guide below.
- Combine palladium trifluoroacetate, triphenylphosphine, sodium carbonate, TFBen, trifluoroethylimidoyl chloride, and amine in an organic solvent like dioxane.
- Heat the reaction mixture to 110 °C and stir for 16 to 30 hours to allow the carbonylation and cyclization to proceed.
- Filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final 2-trifluoromethyl-substituted quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route presents compelling economic and logistical benefits that directly impact the bottom line. The shift away from expensive, pre-activated substrates towards commodity chemicals like trifluoroethylimidoyl chloride and simple amines drastically reduces the raw material cost basis. Furthermore, the elimination of hazardous gaseous carbon monoxide cylinders simplifies facility requirements and safety protocols, lowering the overhead costs associated with specialized handling and storage infrastructure. The operational simplicity of the method, characterized by a one-pot tandem reaction, reduces labor hours and energy consumption compared to multi-step traditional sequences, translating into significant cost reduction in pharmaceutical intermediate manufacturing.
- Cost Reduction in Manufacturing: The economic model of this process is strengthened by the use of commercially available and inexpensive reagents, such as palladium trifluoroacetate and triphenylphosphine, which are used in catalytic amounts. The avoidance of costly coupling agents like T3P and the use of a solid CO surrogate (TFBen) eliminate the need for specialized gas handling equipment, leading to substantial capital expenditure savings. Additionally, the high conversion rates observed across various substrates minimize material waste, ensuring that the cost per kilogram of the final API intermediate is optimized for competitive market positioning.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the reliance on widely available starting materials that are not subject to the same geopolitical or logistical constraints as exotic reagents. The robustness of the reaction conditions allows for flexible scheduling and batch processing, reducing the risk of production delays caused by sensitive reaction parameters. This reliability ensures a consistent flow of high-quality intermediates, enabling downstream drug manufacturers to maintain their production schedules without interruption, thereby securing the continuity of supply for critical medication pipelines.
- Scalability and Environmental Compliance: The method has been demonstrated to be effective on a gram scale with clear potential for expansion to industrial tonnage, addressing the critical need for commercial scale-up of complex pharmaceutical intermediates. The use of dioxane as a solvent, while requiring recovery systems, is a well-established industrial practice, and the solid nature of the CO source simplifies waste management compared to gas scrubbing systems. This alignment with green chemistry principles facilitates easier regulatory approval and environmental compliance, reducing the long-term liability and disposal costs associated with chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity for technical teams evaluating this process for adoption. Understanding these nuances is essential for assessing the feasibility of integrating this method into existing manufacturing workflows.
Q: What is the carbon monoxide source in this novel synthesis method?
A: The method utilizes TFBen (1,3,5-tricarboxylic acid phenol ester) as a solid carbon monoxide substitute, which releases CO under heating conditions, eliminating the need for hazardous gaseous CO handling.
Q: What is the substrate compatibility of this palladium-catalyzed reaction?
A: The reaction demonstrates excellent substrate compatibility, tolerating various functional groups including halogens (F, Cl, Br), alkyl groups (methyl, t-butyl), and trifluoromethyl groups on the aromatic ring.
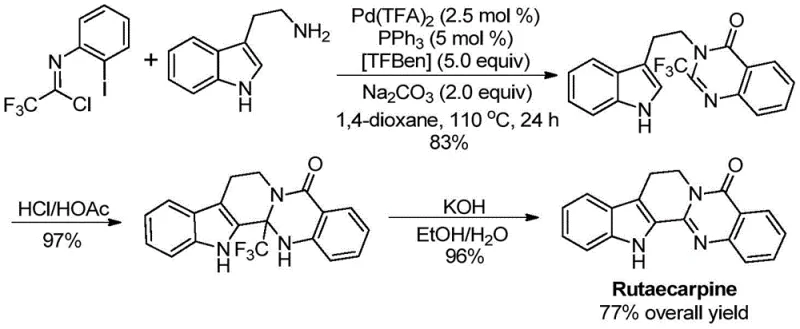
Q: Can this method be applied to the synthesis of complex drug molecules?
A: Yes, the patent explicitly demonstrates the successful application of this method in the high-yield total synthesis of the bioactive natural product Rutaecarpine with a 77% overall yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed carbonylation technology for the next generation of therapeutic agents. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory discovery to market supply is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone intermediate delivered meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this advanced synthetic route for your specific drug development projects. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating exactly how this method can optimize your budget. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you accelerate your timeline to market with confidence and precision.