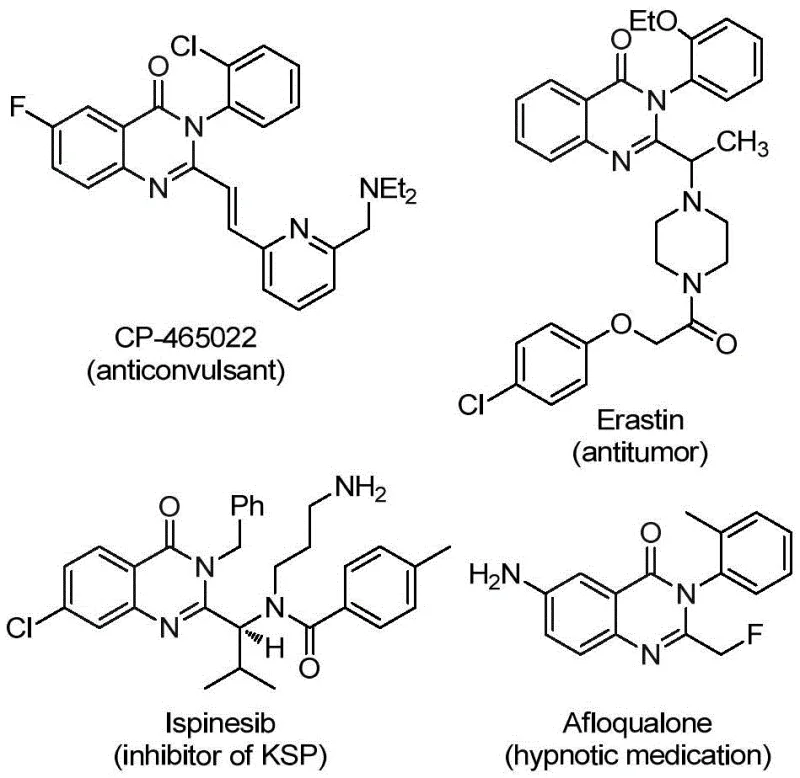
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Commercial Scale-up
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinone Derivatives for Commercial Scale-up
The pharmaceutical industry continuously seeks robust synthetic methodologies to access complex heterocyclic scaffolds that serve as the backbone for next-generation therapeutics. Among these, quinazolinone derivatives represent a privileged structural motif found in numerous bioactive compounds exhibiting anti-inflammatory, antiviral, antifungal, and anticancer properties. The strategic introduction of a trifluoromethyl group at the 2-position of the quinazolinone ring further enhances these pharmacological profiles by improving metabolic stability and lipophilicity. A groundbreaking approach detailed in patent CN112125856A offers a transformative solution for accessing these valuable intermediates. This technology leverages a transition metal palladium-catalyzed carbonylation tandem reaction, utilizing readily available starting materials to construct the core scaffold with high efficiency. By replacing hazardous gaseous carbon monoxide with a solid surrogate, this method addresses critical safety and scalability concerns inherent in traditional carbonylation processes, positioning it as a superior choice for industrial applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-trifluoromethyl substituted quinazolinone derivatives has been fraught with significant technical and operational challenges that hinder large-scale production. Conventional routes often rely on the cyclization of anthranilamides with ethyl trifluoroacetate or trifluoroacetic anhydride, reactions that typically require harsh conditions and expensive, pre-activated substrates. Another common pathway involves the use of unstable trifluoroacetamides or isatoic anhydrides, which suffer from poor atom economy and limited substrate scope. Furthermore, many established protocols necessitate the direct use of carbon monoxide gas, a highly toxic and flammable reagent that demands specialized high-pressure equipment and rigorous safety protocols, drastically increasing capital expenditure and operational risk. These factors collectively result in low yields, narrow functional group tolerance, and prohibitive costs, making traditional methods unsuitable for the reliable supply of high-purity pharmaceutical intermediates required by modern drug discovery pipelines.
The Novel Approach
In stark contrast, the methodology disclosed in the referenced patent introduces a streamlined, palladium-catalyzed strategy that circumvents these historical bottlenecks through innovative reagent selection and mechanistic design. The core innovation lies in the utilization of 1,3,5-tricarboxylate phenol ester (TFBen) as a safe, solid carbon monoxide substitute, which decomposes under heating to release CO in situ, thereby eliminating the need for gas cylinders and high-pressure reactors. This approach couples inexpensive o-iodoanilines with trifluoroethylimidoyl chlorides in a tandem sequence that efficiently constructs the quinazolinone core. The reaction operates under relatively mild conditions in standard organic solvents like tetrahydrofuran, demonstrating exceptional compatibility with a diverse array of substituents including halogens, alkyls, and nitro groups. This versatility allows for the rapid generation of structural analogs, facilitating structure-activity relationship studies while ensuring a cost-effective pathway for commercial manufacturing.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Tandem Reaction
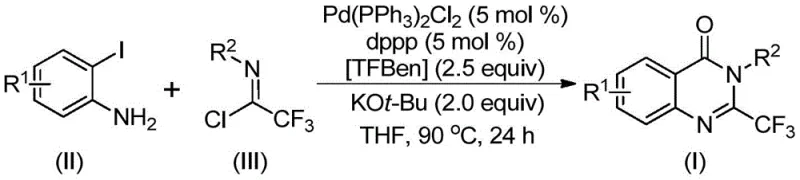
The efficacy of this synthesis is rooted in a sophisticated catalytic cycle that orchestrates multiple bond-forming events in a single pot. The process likely initiates with a base-promoted intermolecular carbon-nitrogen bond coupling between the o-iodoaniline and the trifluoroethylimidoyl chloride, generating a trifluoroacetamidine intermediate. Subsequently, the palladium catalyst, specifically bis(triphenylphosphine)palladium(II) dichloride coordinated with a diphosphine ligand like dppp, undergoes oxidative insertion into the carbon-iodine bond of the aromatic ring. This forms a key divalent palladium species that is poised for carbonyl insertion. As the reaction temperature reaches 90°C, the solid CO source TFBen thermally decomposes, releasing carbon monoxide which inserts into the carbon-palladium bond to form an acyl-palladium intermediate. This step is crucial for introducing the carbonyl functionality required for the lactam ring closure.
Following carbonyl insertion, the presence of a strong base such as potassium tert-butoxide facilitates the deprotonation and subsequent intramolecular nucleophilic attack by the nitrogen atom onto the acyl-palladium center. This cyclization event generates a seven-membered ring palladium intermediate, which then undergoes reductive elimination to release the final 2-trifluoromethyl substituted quinazolinone product and regenerate the active palladium catalyst. This mechanistic pathway not only ensures high atom economy but also minimizes the formation of side products, leading to cleaner reaction profiles. The ability to tolerate various electronic environments on the aromatic rings suggests that the oxidative addition and reductive elimination steps are well-balanced, preventing catalyst deactivation even with electron-deficient or sterically hindered substrates. Such mechanistic robustness is essential for maintaining consistent quality and yield during the commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
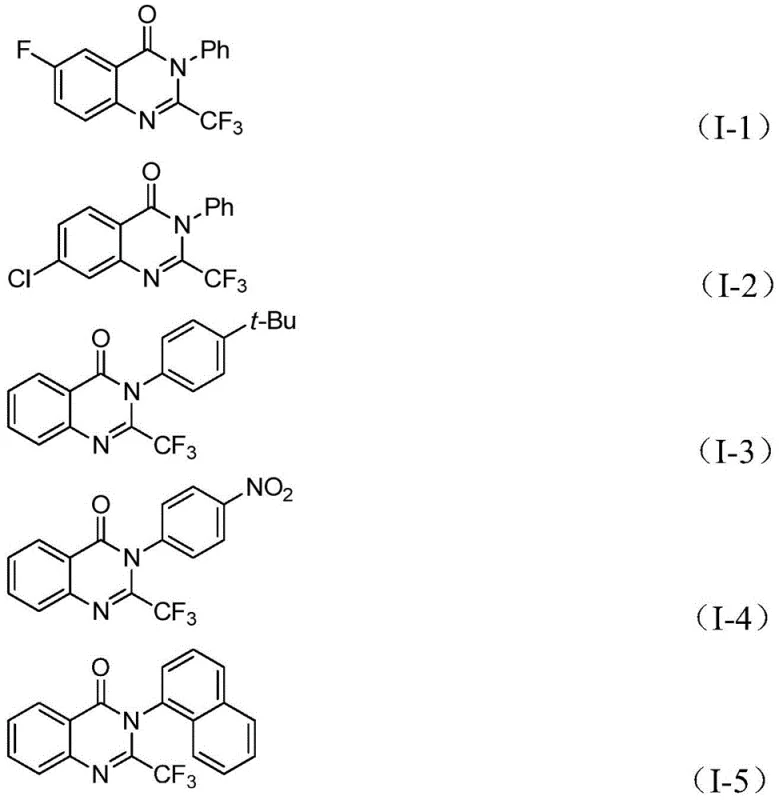
The practical implementation of this synthesis route is designed for ease of operation, requiring standard laboratory glassware and commercially available reagents. The procedure involves charging a reaction vessel with the palladium catalyst system, the solid CO surrogate, the base, and the two primary coupling partners in an aprotic solvent. The mixture is then heated to facilitate the tandem reaction sequence. Detailed standardized synthesis steps, including precise stoichiometric ratios, solvent volumes, and workup procedures optimized for maximum recovery, are outlined in the technical guide below. This protocol has been validated across multiple examples, consistently delivering high purity products suitable for downstream medicinal chemistry applications.
- Combine palladium catalyst, ligand, solid CO substitute (TFBen), base, trifluoroethylimidoyl chloride, and o-iodoaniline in an organic solvent.
- Heat the reaction mixture to 90°C and maintain stirring for 16 to 30 hours to allow the carbonylation tandem reaction to proceed.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers substantial strategic benefits that extend beyond mere chemical yield. The shift from gaseous carbon monoxide to a solid surrogate fundamentally alters the risk profile of the manufacturing process, removing the need for specialized high-pressure infrastructure and reducing regulatory compliance burdens associated with toxic gas handling. This simplification translates directly into lower capital investment requirements and reduced operational overhead, enabling more flexible production scheduling and faster response times to market demands. Furthermore, the use of cheap and readily available starting materials like o-iodoanilines and trifluoroethylimidoyl chlorides ensures a stable supply chain that is less susceptible to raw material price volatility.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the elimination of expensive pre-activated substrates and the avoidance of hazardous gas logistics. By utilizing a solid CO source, manufacturers can operate in standard reactor vessels, significantly lowering equipment maintenance and safety monitoring costs. Additionally, the high catalytic efficiency and broad substrate scope minimize waste generation and purification costs, contributing to a leaner overall production budget. The ability to achieve high conversion rates with inexpensive reagents ensures that the cost per kilogram of the final API intermediate is significantly optimized compared to legacy methods.
- Enhanced Supply Chain Reliability: Reliability in the supply of critical intermediates is paramount for uninterrupted drug production. This method relies on commodity chemicals that are widely sourced from the global chemical market, reducing dependency on niche suppliers. The robustness of the reaction conditions means that batch-to-batch variability is minimized, ensuring consistent quality and delivery timelines. By mitigating the risks associated with toxic gas transport and storage, facilities can maintain continuous operation without the interruptions often caused by safety inspections or regulatory hurdles related to hazardous materials.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the plant floor often reveals hidden inefficiencies, but this protocol is inherently designed for scalability. The use of a solid CO source simplifies the engineering controls required for scale-up, as there is no need for complex gas feed systems. Moreover, the reaction generates fewer hazardous byproducts, aligning with increasingly stringent environmental regulations and sustainability goals. The simplified workup procedure, involving filtration and standard chromatography, facilitates easier isolation of the product, reducing solvent consumption and waste disposal costs associated with large-scale manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for potential partners. Understanding these details is crucial for evaluating the feasibility of integrating this method into existing production workflows.
Q: What are the safety advantages of this synthesis method compared to traditional carbonylation?
A: This method utilizes 1,3,5-tricarboxylate phenol ester (TFBen) as a solid carbon monoxide substitute, effectively eliminating the need for handling toxic and hazardous carbon monoxide gas, thereby significantly enhancing operational safety.
Q: Does this protocol support a wide range of substrate substituents?
A: Yes, the method demonstrates excellent substrate compatibility, accommodating various substituents such as halogens, alkyl groups, and nitro groups on both the aniline and imidoyl chloride components without significant loss in yield.
Q: Why is the introduction of a trifluoromethyl group beneficial in drug design?
A: Incorporating a trifluoromethyl group significantly improves the physicochemical properties of the parent molecule, including enhanced metabolic stability, lipophilicity, and bioavailability, which are critical for developing effective pharmaceutical agents.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development and ensuring supply security. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless and efficient. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. Our expertise in palladium-catalyzed transformations allows us to optimize this specific quinazolinone synthesis for maximum yield and minimal impurity profiles, providing our clients with a competitive edge in the marketplace.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality standards. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation and quality can support your long-term strategic goals in the pharmaceutical sector.