Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
The pharmaceutical and agrochemical industries are constantly seeking robust synthetic routes to access complex heterocyclic scaffolds that serve as critical building blocks for next-generation therapeutics. Among these, quinazolinone derivatives stand out due to their profound biological activities, ranging from antifungal and antiviral properties to potent anticancer effects. A significant breakthrough in this domain is detailed in patent CN112480015B, which discloses a highly efficient multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones. This technology addresses long-standing challenges in heterocyclic chemistry by leveraging a palladium-catalyzed carbonylation cascade that operates under relatively mild conditions. The introduction of the trifluoromethyl group is particularly strategic, as it enhances the metabolic stability, lipophilicity, and bioavailability of the parent molecule, making these intermediates invaluable for drug discovery programs targeting resistant pathogens and oncological indications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core has relied on methodologies that impose significant logistical and safety burdens on manufacturing facilities. Traditional synthetic pathways often necessitate the use of high-pressure carbon monoxide gas, which requires specialized autoclaves and rigorous safety protocols to prevent leakage and exposure. Furthermore, many established routes depend on expensive, pre-activated starting materials such as 2-bromoformylaniline or acid anhydrides, which drive up the raw material costs substantially. Other methods utilizing ruthenium or platinum catalysts often suffer from narrow substrate scope and moderate yields, limiting their utility in diverse medicinal chemistry campaigns. The need for harsh reaction conditions and the generation of stoichiometric metal waste further complicate the purification process, creating bottlenecks in the supply chain for high-purity active pharmaceutical ingredients.
The Novel Approach
In stark contrast, the methodology described in patent CN112480015B offers a streamlined, cost-effective alternative that bypasses these traditional hurdles. By employing a palladium-catalyzed system with molybdenum hexacarbonyl [Mo(CO)6] serving as a solid, safe carbon monoxide surrogate, the process eliminates the need for handling toxic CO gas directly. The reaction utilizes readily available nitro compounds and trifluoroethylimidoyl chloride as starting materials, which are not only cheaper but also exhibit superior atom economy. This one-pot strategy allows for the simultaneous reduction of the nitro group, amidation, and cyclization in a single vessel, drastically reducing processing time and solvent consumption. The operational simplicity, combined with the ability to tolerate a wide range of functional groups, positions this technology as a superior choice for the commercial scale-up of complex pharmaceutical intermediates.
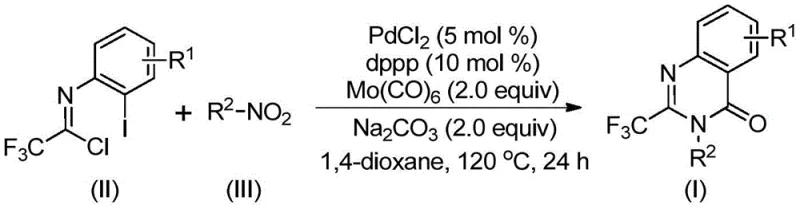
The core transformation involves the reaction of a trifluoroethylimidoyl chloride derivative with a nitro compound in the presence of a palladium catalyst, a phosphine ligand, and a base. As illustrated in the reaction scheme above, the process efficiently constructs the quinazolinone ring system while incorporating the trifluoromethyl group at the 2-position. This general protocol has been validated across a broad spectrum of substrates, demonstrating remarkable versatility. The use of 1,4-dioxane as the solvent and sodium carbonate as the base provides an optimal environment for the catalytic cycle to proceed with high turnover numbers. This level of efficiency is critical for manufacturers aiming to achieve cost reduction in API manufacturing without compromising on the quality or purity of the final output.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cascade
Understanding the mechanistic underpinnings of this transformation is essential for R&D directors looking to optimize the process for specific analogs. The reaction is believed to initiate with the reduction of the nitro compound to the corresponding amine by Mo(CO)6, which simultaneously releases carbon monoxide in situ. This generated amine then undergoes a base-promoted intermolecular coupling with the trifluoroethylimidoyl chloride to form a trifluoroacetamidine intermediate. Subsequently, the palladium catalyst inserts into the carbon-iodine bond of the imidoyl chloride moiety, forming a divalent palladium species. The carbon monoxide released earlier then inserts into the carbon-palladium bond, generating an acyl-palladium intermediate. This key step is followed by an intramolecular nucleophilic attack by the nitrogen atom, facilitated by the base, leading to the formation of a seven-membered palladacycle. Finally, reductive elimination releases the desired 2-trifluoromethyl substituted quinazolinone product and regenerates the active palladium catalyst, closing the catalytic loop.
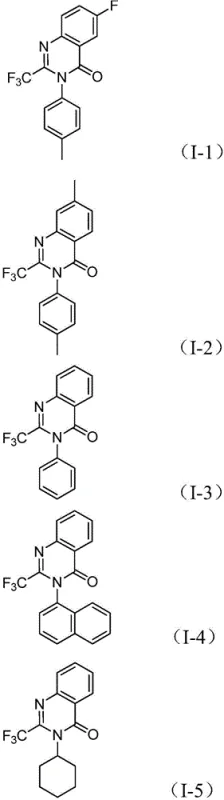
This intricate cascade highlights the importance of ligand selection, specifically 1,3-bis(diphenylphosphino)propane (dppp), which stabilizes the palladium center and facilitates the necessary oxidative addition and reductive elimination steps. The compatibility of this mechanism with various electronic environments on the aromatic rings is a testament to its robustness. Whether the substrate contains electron-withdrawing groups like fluorine or chlorine, or electron-donating groups like methyl or methoxy, the catalytic system maintains high activity. This broad substrate tolerance ensures that medicinal chemists can rapidly generate libraries of analogs for structure-activity relationship (SAR) studies. The ability to synthesize diverse structures, such as those shown below, underscores the utility of this method as a platform technology for developing high-purity OLED material precursors or advanced agrochemical intermediates.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent ratios and reaction parameters to maximize yield and minimize impurities. The standard protocol involves charging a reaction vessel with the palladium catalyst, ligand, base, CO source, and substrates in an anhydrous organic solvent. The mixture is then heated to facilitate the multi-step cascade. While the general procedure is robust, fine-tuning the stoichiometry of the nitro compound relative to the imidoyl chloride can further drive the reaction to completion. For detailed operational specifics regarding temperature ramps, addition sequences, and quenching procedures, please refer to the standardized synthesis guide provided below.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like 1,4-dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours to allow for the carbonylation cascade and cyclization to occur.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers tangible strategic benefits that extend beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the raw material portfolio. By shifting from hazardous high-pressure gases and exotic pre-activated intermediates to stable, commodity-grade nitro compounds and solid Mo(CO)6, companies can significantly de-risk their supply chains. This change reduces dependency on specialized gas suppliers and minimizes the regulatory burden associated with storing and transporting toxic compressed gases. Furthermore, the one-pot nature of the reaction consolidates multiple synthetic steps into a single operation, which inherently reduces labor costs, energy consumption, and solvent waste. These factors collectively contribute to a more resilient and cost-efficient manufacturing model.
- Cost Reduction in Manufacturing: The elimination of high-pressure equipment requirements represents a substantial capital expenditure saving. Traditional carbonylation reactions often demand expensive autoclaves rated for high pressures, whereas this method proceeds effectively in standard glass-lined reactors or stainless steel vessels at atmospheric or slightly elevated pressure. Additionally, the use of inexpensive nitro compounds as starting materials, compared to costly bromo-anilines or anhydrides, lowers the direct material cost per kilogram of product. The high atom economy and reduced number of isolation steps further diminish the overall cost of goods sold (COGS), allowing for more competitive pricing in the global market for fine chemical intermediates.
- Enhanced Supply Chain Reliability: Sourcing reliability is paramount for continuous production schedules. The reagents utilized in this process, including palladium chloride, dppp, and various nitroarenes, are widely available from multiple global chemical suppliers, preventing single-source bottlenecks. The stability of the solid carbon monoxide source, Mo(CO)6, ensures that production is not subject to the delivery logistics and safety constraints of gaseous CO cylinders. This stability allows for larger batch sizes and longer campaign runs without interruption, ensuring a steady flow of intermediates to downstream API synthesis units. Consequently, lead times for high-purity pharmaceutical intermediates can be significantly shortened, enhancing responsiveness to market demands.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method aligns well with modern green chemistry principles. The avoidance of toxic CO gas mitigates the risk of acute exposure incidents, creating a safer working environment for operators. The simplified workup procedure, typically involving filtration and standard chromatography, reduces the volume of aqueous waste streams generated during quenching and extraction. As regulatory scrutiny on chemical manufacturing intensifies, adopting processes with lower hazard profiles and reduced waste generation facilitates easier permitting and compliance with environmental regulations. This scalability ensures that the transition from gram-scale R&D to multi-ton commercial production is seamless and sustainable.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific projects, we have compiled answers to common inquiries regarding the process parameters and scope. These insights are derived directly from the experimental data and embodiments disclosed in the patent literature, providing a reliable foundation for process development decisions. Understanding these nuances helps in planning pilot runs and estimating resource requirements accurately.
Q: What are the key advantages of this one-pot synthesis method over traditional routes?
A: This method eliminates the need for high-pressure carbon monoxide gas and expensive pre-activated substrates. It utilizes cheap nitro compounds and solid Mo(CO)6 as a safe CO source, significantly simplifying the operational setup and reducing safety risks associated with toxic gas handling.
Q: What is the substrate scope for the R2 group in the nitro compound?
A: The process demonstrates excellent compatibility with various substituents. The R2 group can be C1-C10 alkyl, cycloalkyl, or substituted/unsubstituted aryl groups. Specific examples include phenyl, naphthyl, cyclohexyl, and aryl groups with electron-donating or withdrawing groups like methyl, methoxy, or halogens.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the method can be expanded to the gram level and beyond. The use of commercially available reagents, standard solvents like dioxane, and a straightforward workup procedure involving filtration and chromatography makes it highly amenable to commercial scale-up for API intermediate manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development timelines. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be translated into viable industrial processes. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, which utilize state-of-the-art analytical instrumentation to verify identity and assay. Whether you require custom synthesis of novel quinazolinone analogs or bulk supply of established intermediates, our infrastructure is designed to support your growth from early-stage research to full-scale commercialization.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. By partnering with our technical procurement team, you can obtain a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality standards. We encourage potential partners to contact us directly to request specific COA data and route feasibility assessments for your target molecules. Let us help you optimize your supply chain and reduce time-to-market with our reliable, high-quality chemical solutions.