Advanced Pd-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Scale-Up
Advanced Pd-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Scale-Up
The strategic incorporation of trifluoromethyl groups into heterocyclic scaffolds represents a cornerstone of modern medicinal chemistry, profoundly enhancing the metabolic stability, lipophilicity, and bioavailability of drug candidates. As highlighted in patent CN111423381B, a novel and highly efficient preparation method for 2-trifluoromethyl substituted imidazole compounds has been developed, addressing critical bottlenecks in the synthesis of these valuable nitrogen-containing heterocycles. This technology leverages a sophisticated palladium-catalyzed carbonylation strategy that operates under remarkably mild conditions, specifically at 30°C, utilizing safe and accessible reagents to construct the imidazole core with high precision. The significance of this breakthrough extends beyond academic interest, offering a robust pathway for the reliable pharmaceutical intermediate supplier seeking to optimize their production pipelines for high-value bioactive molecules.
The versatility of the resulting 2-trifluoromethyl imidazole derivatives is exemplified by their presence in numerous biologically active architectures, ranging from histamine receptor antagonists to complex coordination ligands used in catalysis. 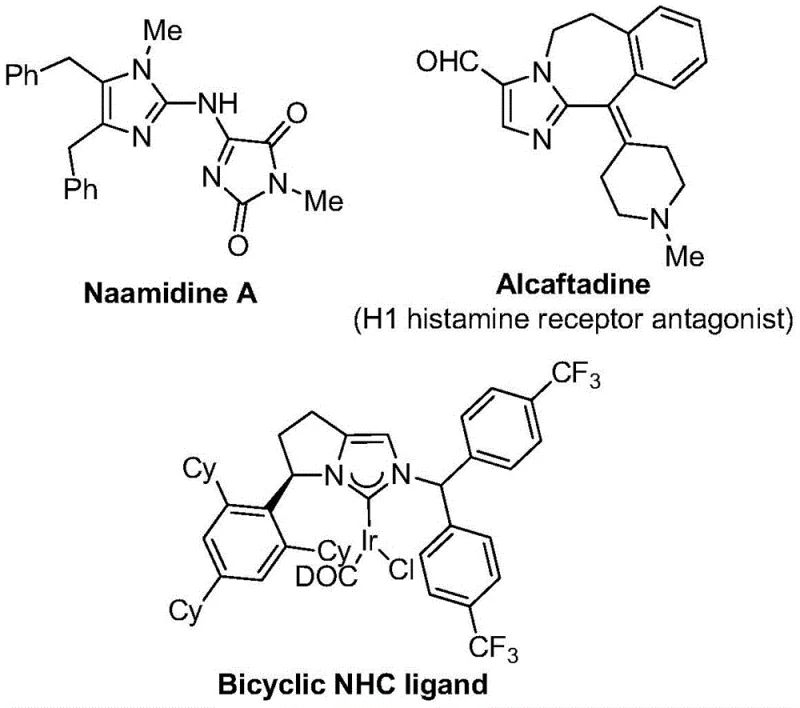 . Traditional synthetic routes often struggle with the introduction of the trifluoromethyl moiety due to the harsh conditions required or the instability of fluorinated synthons. However, this patented methodology circumvents these issues by employing trifluoroethylimidoyl chloride as a stable and reactive synthon, coupled with propargylamine and diaryl iodonium salts. This approach not only simplifies the synthetic sequence but also broadens the applicability of the method across a wide array of substrate designs, making it an indispensable tool for the cost reduction in pharmaceutical intermediate manufacturing.
. Traditional synthetic routes often struggle with the introduction of the trifluoromethyl moiety due to the harsh conditions required or the instability of fluorinated synthons. However, this patented methodology circumvents these issues by employing trifluoroethylimidoyl chloride as a stable and reactive synthon, coupled with propargylamine and diaryl iodonium salts. This approach not only simplifies the synthetic sequence but also broadens the applicability of the method across a wide array of substrate designs, making it an indispensable tool for the cost reduction in pharmaceutical intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted nitrogen heterocycles has been plagued by significant operational challenges that hinder industrial adoption. Conventional methods frequently rely on the direct reaction of unstable trifluoromethyl synthons, such as trifluorodiazoethane, which pose severe safety risks due to their explosive nature and require specialized handling equipment. Furthermore, many existing protocols necessitate the use of toxic carbon monoxide gas under high pressure and elevated temperatures, creating substantial barriers for safe commercial scale-up of complex polymer additives or pharmaceutical ingredients. The limited substrate compatibility of these older methods often results in poor yields when functionalized aryl groups are present, forcing chemists to employ lengthy protection-deprotection sequences that drastically increase production costs and waste generation. Additionally, the reliance on expensive or difficult-to-prepare catalysts in traditional routes further exacerbates the economic inefficiency, making the final high-purity OLED material or API intermediate prohibitively expensive for widespread application.
The Novel Approach
In stark contrast, the innovative process disclosed in the patent utilizes a transition metal palladium-catalyzed carbonylation series reaction that elegantly integrates three readily available starting materials into the target imidazole scaffold in a single pot. 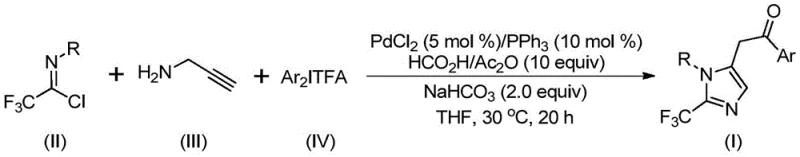 . By replacing hazardous CO gas with a safe in-situ generating system composed of formic acid and acetic anhydride, the method eliminates the need for high-pressure reactors, thereby significantly reducing capital expenditure and safety compliance burdens. The reaction proceeds efficiently at a mild temperature of 30°C in common organic solvents like tetrahydrofuran, demonstrating exceptional energy efficiency compared to thermal cyclizations. Moreover, the use of cheap and easily obtained trifluoroethylimidoyl chloride and diaryl iodonium salts ensures a steady supply chain, while the palladium catalyst system (PdCl2/PPh3) offers high turnover numbers, minimizing metal residue in the final product and simplifying downstream purification processes for the end user.
. By replacing hazardous CO gas with a safe in-situ generating system composed of formic acid and acetic anhydride, the method eliminates the need for high-pressure reactors, thereby significantly reducing capital expenditure and safety compliance burdens. The reaction proceeds efficiently at a mild temperature of 30°C in common organic solvents like tetrahydrofuran, demonstrating exceptional energy efficiency compared to thermal cyclizations. Moreover, the use of cheap and easily obtained trifluoroethylimidoyl chloride and diaryl iodonium salts ensures a steady supply chain, while the palladium catalyst system (PdCl2/PPh3) offers high turnover numbers, minimizing metal residue in the final product and simplifying downstream purification processes for the end user.
Mechanistic Insights into Pd-Catalyzed Carbonylative Cyclization
The mechanistic pathway of this transformation involves a intricate sequence of organometallic steps that ensure high regioselectivity and yield. Initially, an intermolecular carbon-nitrogen bond is formed under alkaline promotion to generate a trifluoroacetamidine intermediate, which subsequently undergoes isomerization. The palladium catalyst then activates the alkyne moiety of the propargylamine through palladation, forming a key alkenyl palladium species. This intermediate undergoes a crucial isomerization to an alkyl palladium complex, positioning the molecule for the subsequent carbonylation event. The carbon monoxide, released slowly from the formic acid/acetic anhydride mixture, inserts into the palladium-carbon bond to create an acyl palladium intermediate. Finally, the diaryl iodonium salt participates via oxidative addition to form a tetravalent palladium species, which undergoes reductive elimination to release the final 2-trifluoromethyl-substituted imidazole compound and regenerate the active catalyst. This detailed understanding of the catalytic cycle allows for precise optimization of reaction parameters to maximize efficiency.
From an impurity control perspective, the mild reaction conditions play a pivotal role in maintaining a clean reaction profile. The low temperature of 30°C suppresses side reactions such as polymerization of the alkyne or decomposition of the sensitive trifluoromethyl group, which are common pitfalls in high-temperature syntheses. The specific choice of sodium bicarbonate as a base ensures neutralization of acidic byproducts without promoting unwanted nucleophilic attacks on the electrophilic centers of the intermediates. Furthermore, the stoichiometric balance of the reagents, particularly the slight excess of trifluoroethylimidoyl chloride relative to propargylamine, drives the reaction to completion while minimizing the formation of homocoupling byproducts from the iodonium salt. This inherent selectivity reduces the burden on purification teams, ensuring that the reducing lead time for high-purity pharmaceutical intermediates is achieved without compromising on the stringent quality standards required for GMP manufacturing environments.
How to Synthesize 2-Trifluoromethyl Imidazole Efficiently
The practical execution of this synthesis is designed for simplicity and reproducibility, making it ideal for both laboratory discovery and pilot plant operations. The protocol involves charging a reaction vessel with the palladium catalyst system, the CO source additives, and the three key substrates in a suitable aprotic solvent. The detailed standardized synthesis steps see the guide below, which outlines the precise molar ratios and addition sequences required to achieve optimal conversion rates exceeding 90% in many cases.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, and the CO source system (formic acid/acetic anhydride) in an organic solvent like THF.
- Add the substrates: trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 16-24 hours, then filter and purify via column chromatography to isolate the target imidazole.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, this patented technology offers a compelling value proposition centered around cost efficiency and supply security. The shift from hazardous gaseous reagents to stable liquid or solid precursors fundamentally alters the risk profile of the manufacturing process, leading to lower insurance premiums and reduced regulatory overhead. The ability to source starting materials like propargylamine and various aromatic amines from bulk chemical suppliers ensures a resilient supply chain that is less susceptible to geopolitical disruptions or single-source bottlenecks. Furthermore, the high atom economy of the carbonylation reaction minimizes waste disposal costs, aligning with increasingly strict environmental regulations and corporate sustainability goals. This holistic improvement in process economics translates directly into a more competitive pricing structure for the final active pharmaceutical ingredients or fine chemical intermediates.
- Cost Reduction in Manufacturing: The elimination of high-pressure carbon monoxide infrastructure and the use of inexpensive palladium chloride instead of exotic ligands result in substantial capital and operational expenditure savings. By avoiding cryogenic conditions or extreme heating, the energy consumption per kilogram of product is drastically lowered, contributing to a leaner manufacturing budget. The high yields reported across a broad substrate scope mean that less raw material is wasted, further driving down the cost of goods sold (COGS) and improving overall margin potential for the manufacturing partner.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as formic acid, acetic anhydride, and sodium bicarbonate ensures that production can continue uninterrupted even during fluctuations in the specialty chemical market. The robustness of the reaction to various functional groups allows for the flexible sourcing of different aryl building blocks, providing procurement teams with multiple vendor options for raw materials. This flexibility mitigates the risk of production stoppages due to raw material shortages, ensuring consistent delivery schedules to downstream customers who depend on just-in-time inventory models.
- Scalability and Environmental Compliance: The mild reaction conditions and simple workup procedure involving filtration and column chromatography facilitate seamless scale-up from gram to ton quantities without the need for complex process re-engineering. The absence of toxic heavy metal residues in the final product, thanks to the efficient catalytic cycle, simplifies the purification process and ensures compliance with strict limits on residual metals in pharmaceutical products. Additionally, the use of safer reagents reduces the generation of hazardous waste streams, lowering the environmental footprint and associated disposal costs, which is a critical factor for long-term sustainable operations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route, derived directly from the patent specifications and experimental data. These insights are intended to clarify the operational feasibility and strategic benefits for potential partners evaluating this technology for their own production portfolios.
Q: What are the key advantages of this Pd-catalyzed method over traditional imidazole synthesis?
A: This method operates under exceptionally mild conditions (30°C) compared to traditional high-temperature cyclizations. It utilizes a safe in-situ CO source (formic acid/acetic anhydride) rather than toxic carbon monoxide gas, significantly enhancing operational safety and reducing equipment costs for industrial scale-up.
Q: Is this synthesis route scalable for large-volume production of pharmaceutical intermediates?
A: Yes, the patent explicitly demonstrates scalability potential. The use of inexpensive catalysts (PdCl2/PPh3) and readily available starting materials like propargylamine and diaryl iodonium salts ensures that the process is economically viable for commercial scale-up from gram to multi-kilogram levels without complex purification bottlenecks.
Q: What is the substrate scope regarding the aryl groups in this reaction?
A: The reaction exhibits excellent functional group tolerance. It successfully accommodates various substituents on both the imidoyl chloride and the iodonium salt, including electron-donating groups like methyl and methoxy, as well as electron-withdrawing groups such as halogens, nitro, and trifluoromethyl, allowing for diverse molecular design.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed methodology for the production of next-generation pharmaceutical intermediates and fine chemicals. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is smooth and efficient. We are committed to delivering high-purity 2-trifluoromethyl imidazole derivatives that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch. Our dedication to quality and consistency makes us the preferred partner for global enterprises seeking to secure their supply of critical heterocyclic building blocks.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits of switching to this greener, more efficient process. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate our capability to support your R&D and commercial manufacturing needs with reliability and expertise.