Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Scale-Up
Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Imidazoles for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to access fluorinated heterocycles, which serve as critical scaffolds in modern drug discovery and material science. Patent CN111423381B introduces a groundbreaking preparation method for 2-trifluoromethyl substituted imidazole compounds, addressing the longstanding challenges associated with introducing trifluoromethyl groups into nitrogen-containing five-membered heterocycles. This technology leverages a transition metal palladium-catalyzed carbonylation series reaction, utilizing cheap and easily obtainable starting materials such as trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts. The significance of this innovation lies in its ability to operate under remarkably mild conditions, specifically at 30°C, thereby minimizing energy consumption and reducing the formation of thermal degradation byproducts. As illustrated in the structural diversity of bioactive molecules below, the imidazole core is ubiquitous in high-value applications ranging from H1 histamine receptor antagonists to complex ligand systems, making efficient access to these derivatives a priority for any reliable pharmaceutical intermediate supplier.
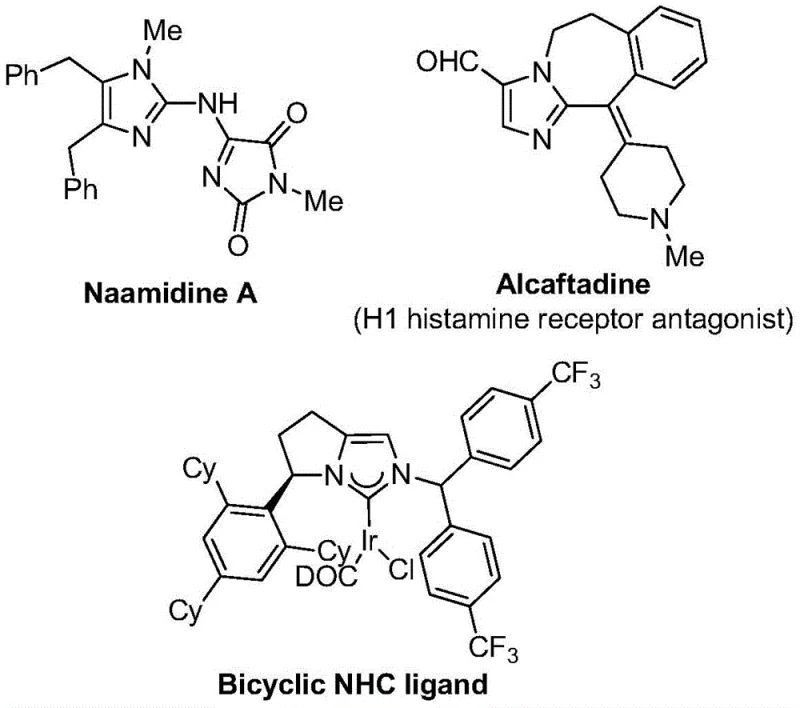
Imidazole compounds are not merely academic curiosities; they are foundational elements in the architecture of countless active pharmaceutical ingredients (APIs) and functional materials. The introduction of a trifluoromethyl group into these heterocyclic systems confers distinct physicochemical advantages, including significantly improved electronegativity, enhanced metabolic stability, and optimized lipophilicity, which are crucial parameters for improving the bioavailability of drug candidates. Traditional synthetic routes often rely on hazardous reagents like trifluorodiazoethane or require harsh reaction conditions that limit substrate scope and pose safety risks during scale-up. In contrast, the methodology disclosed in this patent utilizes trifluoroethylimidoyl halides, which, while historically underutilized, possess immense application potential when paired with the right catalytic system. This novel approach transforms these readily available synthons into high-value 2-trifluoromethyl imidazoles through a streamlined process that is both operationally simple and highly efficient, representing a significant leap forward in cost reduction in pharmaceutical intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nitrogen-containing heterocycles bearing trifluoromethyl functional groups has been fraught with technical and economic hurdles. Conventional literature methods predominantly rely on the direct reaction of trifluoromethyl synthons, such as trifluorodiazoethane, with suitable substrates. While effective in specific contexts, trifluorodiazoethane is inherently unstable and potentially explosive, necessitating specialized handling equipment and stringent safety protocols that drive up operational costs and complicate regulatory compliance. Furthermore, many existing protocols require elevated temperatures or strong bases that can lead to poor functional group tolerance, resulting in complex impurity profiles that are difficult to purge during downstream processing. These limitations severely restrict the commercial scale-up of complex polymer additives or pharmaceutical intermediates derived from these sensitive pathways. Additionally, the reliance on exotic or difficult-to-source reagents often creates supply chain bottlenecks, leading to inconsistent availability and volatile pricing for the final heterocyclic products, which is a major concern for procurement managers aiming for supply chain continuity.
The Novel Approach
The methodology presented in patent CN111423381B offers a transformative solution by employing a palladium-catalyzed multicomponent coupling strategy that circumvents the drawbacks of traditional syntheses. By utilizing trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts as the primary building blocks, this route accesses the desired 2-trifluoromethyl imidazole core through a cascade of well-controlled organometallic transformations. The reaction proceeds efficiently at a mild temperature of 30°C, which not only enhances safety but also preserves sensitive functional groups that would otherwise decompose under harsher conditions. This approach allows for the design and synthesis of diversified substituted imidazole compounds with trifluoromethyl groups at the 1 and 5 positions, providing medicinal chemists with a versatile toolbox for structure-activity relationship (SAR) studies. The use of commercially available and inexpensive catalysts like palladium chloride, combined with common ligands such as triphenylphosphine, ensures that the process remains economically viable for large-scale production, effectively lowering the barrier to entry for high-purity OLED material or agrochemical intermediate synthesis.
Mechanistic Insights into Pd-Catalyzed Carbonylation Cyclization
The success of this synthetic route hinges on a sophisticated catalytic cycle driven by palladium, which orchestrates the assembly of the imidazole ring with high precision. The mechanism initiates with the formation of an intermolecular carbon-nitrogen bond promoted by the alkaline additive, generating a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization, setting the stage for the palladium-catalyzed alkyne amination. The palladium center coordinates with the alkyne moiety of the propargylamine derivative, undergoing palladation to form a key alkenyl palladium intermediate. This intermediate then isomerizes to an alkyl palladium species, which is poised for the critical carbonylation step. Crucially, the carbon monoxide required for this transformation is generated in situ from the decomposition of formic acid and acetic anhydride, eliminating the need for handling toxic CO gas cylinders. The resulting acyl palladium intermediate then engages in an oxidative addition with the diaryl iodonium salt, forming a transient tetravalent palladium species. Finally, reductive elimination releases the final 2-trifluoromethyl-substituted imidazole product and regenerates the active palladium catalyst, completing the cycle. This intricate dance of organometallic steps ensures high atom economy and minimizes waste generation.
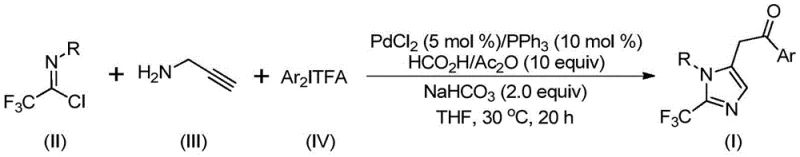
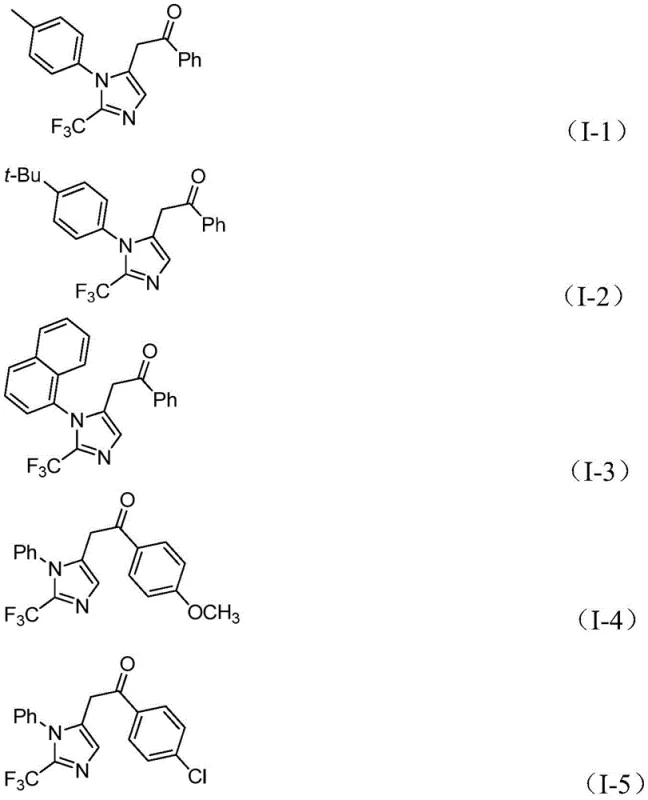
From an impurity control perspective, the mild reaction conditions play a pivotal role in ensuring the quality of the final product. Operating at 30°C significantly reduces the likelihood of thermal decomposition of the reactive intermediates, such as the alkenyl palladium species, which could otherwise lead to polymeric byproducts or oligomerization of the alkyne starting material. Furthermore, the use of sodium bicarbonate as a mild base helps to neutralize acidic byproducts without promoting unwanted side reactions like hydrolysis of the imidoyl chloride or the final ketone functionality. The compatibility of this system with a wide range of substituents, including electron-withdrawing groups like nitro and halogens as well as electron-donating groups like methoxy and tert-butyl, demonstrates the robustness of the catalytic cycle against electronic perturbations. This broad substrate tolerance means that the impurity profile remains consistent and predictable across different analogues, simplifying the purification process and ensuring that the high-purity pharmaceutical intermediates meet stringent regulatory specifications required for clinical applications.
How to Synthesize 2-Trifluoromethyl Imidazoles Efficiently
The practical implementation of this synthesis is designed for ease of execution in both laboratory and pilot plant settings. The protocol involves a straightforward one-pot procedure where all reagents are combined in an aprotic organic solvent, preferably tetrahydrofuran (THF), which has been shown to effectively promote the reaction with high conversion rates. The molar ratios are carefully optimized, typically employing a slight excess of the trifluoroethylimidoyl chloride and diaryl iodonium salt relative to the propargylamine to drive the reaction to completion. The detailed standardized synthesis steps, including precise weighing, addition sequences, and workup procedures involving silica gel filtration and column chromatography, are outlined in the guide below to ensure reproducibility and safety for technical teams looking to adopt this technology.
- Combine palladium chloride, triphenylphosphine, sodium bicarbonate, acetic anhydride, and formic acid in an organic solvent such as THF.
- Add trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt to the reaction mixture under stirring.
- Maintain the reaction at 30°C for 18 to 20 hours, then filter and purify via column chromatography to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers substantial strategic benefits that extend beyond mere chemical efficiency. The reliance on cheap and easily obtained starting materials, such as aromatic amines for the preparation of trifluoroethylimidoyl chloride and commercially available propargylamine, drastically simplifies the raw material sourcing landscape. Unlike processes that depend on custom-synthesized or hazardous reagents, the inputs for this reaction are commodity chemicals with stable global supply chains, reducing the risk of production stoppages due to material shortages. Furthermore, the elimination of expensive transition metal catalysts in favor of relatively inexpensive palladium chloride, used at low loading levels (5 mol%), contributes to significant cost optimization in the overall manufacturing budget. The simplicity of the post-treatment process, which involves standard filtration and chromatography rather than complex distillation or crystallization steps, further reduces operational expenditures related to energy and labor.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of readily available feedstocks and a highly efficient catalytic system that minimizes waste. By avoiding the use of hazardous diazo compounds and high-pressure carbon monoxide gas, the facility saves on specialized safety infrastructure and insurance costs. The mild reaction temperature of 30°C translates to lower energy consumption for heating and cooling compared to traditional reflux conditions, directly impacting the utility costs per kilogram of product. Additionally, the high reaction efficiency and yield reported in the patent examples mean that less raw material is wasted on side products, maximizing the output from every batch and enhancing the overall return on investment for the manufacturing campaign.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the modular nature of this synthesis, which allows for the easy substitution of various aryl groups on both the imidoyl chloride and the iodonium salt components. This flexibility means that if a specific substituent becomes unavailable, the process can be rapidly adapted to use alternative analogues without requiring a complete re-optimization of the reaction conditions. The use of common solvents like THF and standard reagents like sodium bicarbonate ensures that the supply chain is not dependent on niche vendors. This robustness allows for better inventory management and longer-term contracting with suppliers, securing favorable pricing and ensuring a continuous flow of materials necessary for uninterrupted commercial production schedules.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram or even tonne levels is facilitated by the benign reaction conditions and the absence of highly exothermic steps that are difficult to control in large reactors. The generation of carbon monoxide in situ from formic acid and acetic anhydride eliminates the need for storing and transporting large quantities of toxic gas, significantly improving the environmental, health, and safety (EHS) profile of the facility. The waste stream is primarily composed of organic solvents and inorganic salts, which can be managed through standard waste treatment protocols, ensuring compliance with increasingly strict environmental regulations. This ease of scale-up makes the technology ideal for meeting the growing demand for fluorinated heterocycles in the pharmaceutical and agrochemical sectors without compromising on safety or sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis method, derived directly from the experimental data and disclosures within the patent documentation. Understanding these nuances is critical for R&D teams evaluating the feasibility of this route for their specific target molecules. The answers provided reflect the optimized conditions and observed substrate tolerances that define the operational window of this technology.
Q: What are the optimal reaction conditions for this synthesis?
A: The patent specifies a mild temperature of 30°C with a reaction time of 16 to 24 hours, utilizing THF as the preferred solvent and a PdCl2/PPh3 catalyst system.
Q: Can this method tolerate diverse functional groups on the aryl rings?
A: Yes, the method demonstrates excellent substrate compatibility, successfully accommodating substituents such as methyl, tert-butyl, halogens, trifluoromethyl, and nitro groups on both the imidoyl chloride and the iodonium salt.
Q: What is the source of the carbonyl group in the final imidazole structure?
A: The carbonyl functionality is introduced via a carbonylation reaction where carbon monoxide is generated in situ from the decomposition of formic acid and acetic anhydride.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this palladium-catalyzed carbonylation technology for the production of high-value fluorinated intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped to handle the specific requirements of this chemistry, including the safe handling of palladium catalysts and the rigorous purification needed to achieve stringent purity specifications. With our dedicated rigorous QC labs, we guarantee that every batch of 2-trifluoromethyl imidazole delivered meets the highest standards of quality and consistency, supporting your drug development timelines with reliability.
We invite you to collaborate with us to leverage this advanced synthetic route for your next project. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and target specifications. Please contact our technical procurement team today to request specific COA data for our catalog compounds or to discuss route feasibility assessments for your proprietary molecules. Let us help you optimize your supply chain and accelerate your time to market with our superior manufacturing capabilities.