Scalable Synthesis of 2-Trifluoromethyl Quinazolinones Using Inexpensive Iron Catalysis for Commercial API Production
Introduction to Advanced Quinazolinone Synthesis
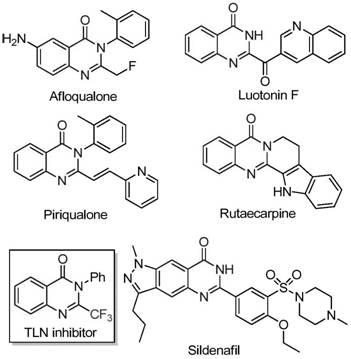
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting potent biological activity. Patent CN111675662A discloses a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone compounds, addressing critical bottlenecks in the production of these valuable pharmacophores. Quinazolinones are ubiquitous in natural products and drug molecules, known for their anticancer, anticonvulsant, anti-inflammatory, and antifungal properties. The strategic introduction of a trifluoromethyl group further enhances these molecules by improving metabolic stability, lipophilicity, and bioavailability. This technical insight report analyzes the novel iron-catalyzed tandem cyclization described in the patent, highlighting its potential to revolutionize the supply chain for reliable pharmaceutical intermediate suppliers seeking cost-effective and scalable solutions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted quinazolinones has relied on methodologies that pose significant challenges for large-scale manufacturing. Traditional literature reports predominantly utilize trifluoromethyl synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate reacting with substrates like anthranilamide or isatoic anhydride. These conventional pathways are frequently plagued by severe reaction conditions that require extreme temperatures or harsh reagents, leading to safety concerns and increased energy consumption. Furthermore, the starting materials involved are often expensive and difficult to source in bulk quantities, creating volatility in the supply chain. Perhaps most critically, these older methods suffer from narrow substrate scope and low yields, limiting their utility for generating diverse compound libraries required in drug discovery pipelines. The reliance on such inefficient processes results in higher production costs and greater environmental waste, which are unacceptable in modern green chemistry standards.
The Novel Approach
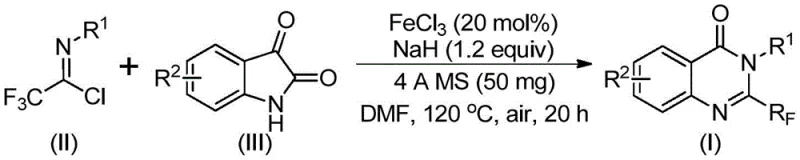
In stark contrast, the methodology outlined in patent CN111675662A introduces a paradigm shift by employing readily available and inexpensive starting materials: trifluoroacetimidoyl chloride and isatin. This novel approach utilizes a tandem cyclization reaction catalyzed by inexpensive metallic iron, specifically ferric chloride, which drastically reduces the cost of goods sold. The reaction proceeds under relatively mild conditions, utilizing a two-stage temperature profile that ensures high conversion rates without degrading sensitive functional groups. By shifting away from precious metal catalysts and expensive fluorinating agents, this method offers a robust alternative for cost reduction in API manufacturing. The process demonstrates exceptional functional group tolerance, accommodating various substituents on the aromatic rings, which allows for the rapid synthesis of a wide array of derivatives. This flexibility is crucial for pharmaceutical companies aiming to optimize lead compounds without being constrained by synthetic limitations.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this technological breakthrough lies in the intricate mechanism facilitated by the iron catalyst and base system. The reaction is proposed to initiate with an alkali-promoted intermolecular carbon-nitrogen bond formation between the trifluoroacetimidoyl chloride and the isatin derivative. This initial step generates a trifluoroacetamidine intermediate, setting the stage for the subsequent ring closure. The presence of ferric chloride is critical, as it likely coordinates with the carbonyl oxygen or the nitrogen atoms to activate the substrate towards cyclization. Following the initial bond formation, the system undergoes an iron-catalyzed decarbonylation process, which is the key step in forming the quinazolinone core while expelling carbon monoxide. Finally, an isomerization step yields the thermodynamically stable 2-trifluoromethyl substituted quinazolinone product. Understanding this mechanistic pathway is vital for R&D directors, as it explains the high selectivity and purity observed in the final products, minimizing the formation of regioisomers or side products that complicate downstream purification.
Furthermore, the impurity control mechanism inherent in this design is superior to traditional acid-mediated cyclizations. The use of sodium hydride as a base ensures complete deprotonation of the isatin nitrogen, driving the reaction forward and preventing the accumulation of unreacted starting materials. The addition of 4A molecular sieves plays a subtle yet important role by scavenging trace moisture, which could otherwise hydrolyze the sensitive imidoyl chloride or deactivate the catalyst. This attention to reaction engineering details ensures that the crude reaction mixture is cleaner, thereby reducing the burden on purification teams. For quality control laboratories, this means a simpler impurity profile to characterize and validate, accelerating the timeline from bench-scale synthesis to GMP production. The ability to predict and control these mechanistic variables provides a significant competitive advantage in maintaining stringent purity specifications required for clinical trial materials.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
The operational simplicity of this synthesis makes it highly attractive for process chemists looking to implement new routes quickly. The protocol involves charging a reactor with the iron catalyst, sodium hydride, molecular sieves, and the two primary substrates in a polar aprotic solvent such as DMF. The reaction is carefully managed through a temperature ramp, starting at a lower temperature to facilitate the initial coupling and then heating to drive the cyclization to completion. Detailed standardized synthesis steps for this specific transformation are provided in the guide below, ensuring reproducibility across different laboratory settings.
- Mix ferric chloride catalyst, sodium hydride, 4A molecular sieves, trifluoroacetimidoyl chloride, and isatin in an organic solvent like DMF.
- Stir the reaction mixture at 40°C for 8 to 10 hours to initiate the intermolecular carbon-nitrogen bond formation.
- Heat the reaction to 120°C for 18 to 20 hours to complete the decarbonylation cyclization, then purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the shift to this iron-catalyzed methodology offers substantial strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the drastic reduction of raw material costs, driven by the substitution of expensive precious metal catalysts with commodity-grade ferric chloride. This change not only lowers the direct material cost but also simplifies the supply chain by reducing dependency on specialized catalyst vendors who may have long lead times. Additionally, the use of isatin and trifluoroacetimidoyl chloride as starting materials leverages widely available chemical feedstocks, ensuring supply continuity even during market fluctuations. For supply chain heads, this reliability is paramount in maintaining uninterrupted production schedules for critical API intermediates.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts removes the need for expensive metal scavenging steps and complex residue testing, which are significant cost drivers in pharmaceutical manufacturing. By utilizing base metal catalysis, the process inherently lowers the cost of goods, allowing for more competitive pricing in the global market. Furthermore, the high yields reported in the patent data suggest that less raw material is wasted per kilogram of product, enhancing overall process efficiency. These factors combine to create a leaner manufacturing model that maximizes margin potential without compromising on quality standards.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route against varying substrate electronics means that a single platform technology can be used to produce multiple analogues. This versatility reduces the need for developing entirely new processes for each new drug candidate, speeding up time-to-market. The availability of starting materials like isatin derivatives from multiple global suppliers mitigates the risk of single-source bottlenecks. Consequently, procurement managers can negotiate better terms and secure long-term contracts with greater confidence, knowing that the underlying chemistry is not dependent on obscure or proprietary reagents.
- Scalability and Environmental Compliance: The reaction conditions described are amenable to scale-up, having been demonstrated effectively at the gram level with potential for ton-scale production. The use of DMF as a solvent, while requiring careful handling, is a standard industrial solvent with established recovery and recycling protocols, facilitating compliance with environmental regulations. The simplified workup procedure, involving filtration and standard chromatography, reduces the generation of hazardous waste compared to multi-step traditional syntheses. This alignment with green chemistry principles not only reduces disposal costs but also enhances the corporate sustainability profile, a key metric for modern pharmaceutical partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method, derived directly from the experimental data and claims within the patent documentation. These insights are intended to clarify the practical implications of adopting this technology for your specific manufacturing needs. Understanding these nuances helps stakeholders make informed decisions about process integration and resource allocation.
Q: What are the advantages of using FeCl3 over traditional catalysts for quinazolinone synthesis?
A: Ferric chloride is significantly cheaper and more abundant than precious metal catalysts often used in heterocycle synthesis. The patent demonstrates that FeCl3 enables high yields (up to 93%) under relatively mild conditions, reducing both raw material costs and waste disposal expenses associated with heavy metals.
Q: Can this synthesis method tolerate diverse functional groups on the substrate?
A: Yes, the method exhibits excellent functional group tolerance. The patent data confirms successful synthesis with substrates containing methyl, fluoro, bromo, chloro, methoxy, and nitro groups at various positions (ortho, meta, para), making it highly versatile for generating diverse libraries of pharmaceutical intermediates.
Q: Is this process suitable for large-scale industrial production?
A: The patent explicitly states that the method is scalable to the gram level and potentially for industrial application. The use of inexpensive reagents like isatin and trifluoroacetimidoyl chloride, combined with a simple workup procedure involving filtration and column chromatography, supports its feasibility for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the iron-catalyzed synthesis described in patent CN111675662A for the production of high-value pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab-scale discovery to full-scale manufacturing is seamless. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone delivered meets the highest industry standards. We are committed to leveraging this advanced chemistry to provide our clients with a competitive edge in the marketplace.
We invite you to engage with our technical procurement team to discuss how this innovative route can be tailored to your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits specific to your volume needs. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate our capability as your trusted partner in delivering high-purity pharmaceutical intermediates efficiently and reliably.