Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Global Pharma Supply Chains
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Global Pharma Supply Chains
The pharmaceutical industry continuously seeks robust and scalable methodologies for constructing privileged heterocyclic scaffolds, particularly those exhibiting potent biological activities. Patent CN112480015A discloses a groundbreaking multicomponent one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones, a class of compounds renowned for their antifungal, antibacterial, antiviral, and anticancer properties. The introduction of a trifluoromethyl group significantly enhances the physicochemical profile of these molecules, improving lipophilicity, metabolic stability, and bioavailability, which are critical parameters in modern drug design. This innovative approach leverages transition metal palladium catalysis to couple trifluoroethylimidoyl chloride with nitro compounds, bypassing the limitations of traditional synthetic routes. The versatility of this scaffold is evident in numerous marketed drugs and clinical candidates, highlighting the immense commercial potential of mastering this specific chemical architecture for reliable pharmaceutical intermediate supplier networks globally.
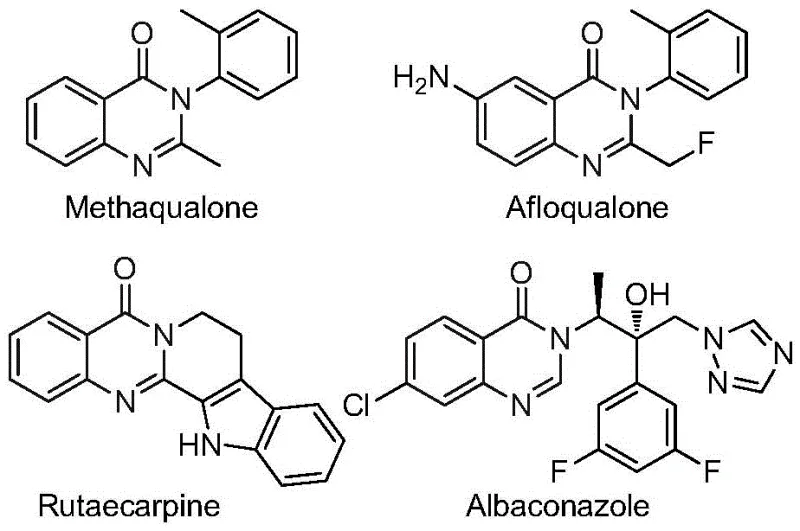
Quinazolinone compounds represent a cornerstone in medicinal chemistry, found in a vast array of functional molecules ranging from fluoroquinolones to specialized kinase inhibitors. The structural diversity achievable within this fused-ring nitrogen-containing six-membered heterocycle system allows for precise tuning of biological activity. As illustrated by the variety of bioactive molecules shown, the core quinazolinone structure serves as a versatile platform for developing new therapeutic agents. The specific incorporation of the trifluoromethyl moiety at the 2-position further amplifies these properties, making the efficient synthesis of these derivatives a high-priority objective for R&D teams focused on oncology and infectious disease treatments. The ability to access these complex structures through a streamlined, one-pot process represents a significant leap forward in process chemistry, offering a pathway to high-purity pharmaceutical intermediates with reduced environmental impact.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of quinazolinone rings has been plagued by significant synthetic challenges that hinder large-scale production and cost efficiency. Traditional methods often rely on the reduction of nitro-substituted benzamides catalyzed by expensive ruthenium or platinum complexes under high-pressure carbon monoxide atmospheres, which poses severe safety risks and requires specialized high-pressure equipment. Other approaches involve iron-catalyzed condensations that may suffer from poor atom economy or palladium-catalyzed cyclizations requiring pre-activated substrates like 2-bromoformanilides or 2-iodoanilines. These conventional routes are frequently limited by harsh reaction conditions, narrow substrate scopes, and the necessity for multiple synthetic steps to prepare the starting materials. Furthermore, the reliance on gaseous carbon monoxide introduces substantial logistical and safety burdens, complicating the commercial scale-up of complex pharmaceutical intermediates and increasing the overall cost of goods sold due to stringent safety protocols and equipment requirements.
The Novel Approach
In stark contrast to these legacy methods, the technology described in patent CN112480015A introduces a highly efficient, palladium-catalyzed carbonylation serial reaction that utilizes cheap and easily obtainable nitro compounds and trifluoroethylimidoyl chloride as starting materials. This novel one-pot strategy eliminates the need for high-pressure CO gas by employing molybdenum hexacarbonyl as a safe, solid carbon monoxide substitute, thereby drastically simplifying the operational setup and enhancing workplace safety. The reaction proceeds smoothly in common organic solvents like dioxane at moderate temperatures, demonstrating exceptional tolerance for a wide range of functional groups including halogens, alkyls, and electron-withdrawing groups. This method not only streamlines the synthesis into a single operational step but also achieves high reaction efficiencies and yields, as evidenced by the successful preparation of various substituted derivatives. By removing the need for pre-activation of substrates and utilizing abundant nitro compounds, this approach offers a compelling solution for cost reduction in pharmaceutical intermediate manufacturing while maintaining high standards of purity and structural integrity.
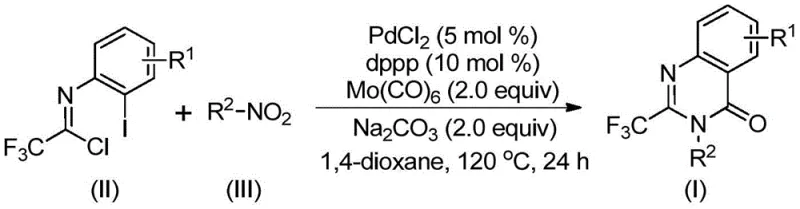
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cyclization
The mechanistic pathway of this transformation is a sophisticated orchestration of reduction, coupling, and cyclization events driven by the palladium catalyst and the carbon monoxide source. Initially, the nitro compound undergoes reduction to the corresponding amine species, likely facilitated by the molybdenum hexacarbonyl under the thermal conditions. This generated amine then participates in an alkali-promoted coupling with the trifluoroethylimidoyl chloride to form a trifluoroacetamidine derivative intermediate. Subsequently, the palladium catalyst inserts into the carbon-iodine bond of the aromatic ring, forming a reactive divalent palladium intermediate. Simultaneously, the molybdenum hexacarbonyl releases carbon monoxide upon heating, which inserts into the carbon-palladium bond to generate an acyl palladium species. This key acyl intermediate then undergoes intramolecular cyclization promoted by the base, forming a seven-membered ring palladium complex before finally undergoing reductive elimination to release the desired 2-trifluoromethyl-substituted quinazolinone product. This intricate cascade ensures high regioselectivity and minimizes the formation of side products, which is crucial for maintaining a clean impurity profile in pharmaceutical synthesis.
Understanding the impurity control mechanisms inherent in this catalytic cycle is vital for ensuring the quality of the final active pharmaceutical ingredient. The use of a specific ligand system, such as 1,3-bis(diphenylphosphino)propane (dppp), alongside palladium chloride, optimizes the electronic and steric environment around the metal center, favoring the desired cyclization pathway over competing side reactions like homocoupling or simple dehalogenation. The stoichiometric balance between the palladium catalyst, the ligand, and the base (sodium carbonate) is critical; the patent specifies a molar ratio that maximizes turnover while minimizing residual metal contamination. Furthermore, the choice of solvent plays a pivotal role in solubilizing the polar nitro compounds and the organometallic intermediates, ensuring a homogeneous reaction environment that promotes consistent kinetics. By carefully controlling these parameters, the process effectively suppresses the formation of difficult-to-remove impurities, thereby reducing the burden on downstream purification processes and enhancing the overall yield of high-purity pharmaceutical intermediates suitable for clinical applications.
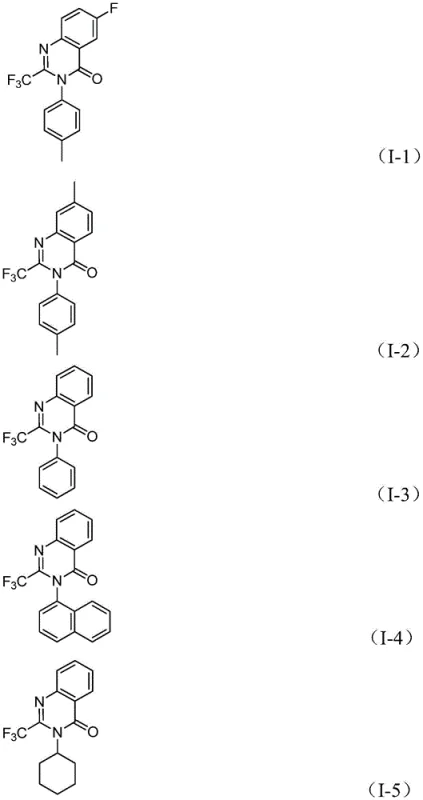
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The practical execution of this synthesis is designed for scalability and ease of operation, making it highly attractive for contract development and manufacturing organizations (CDMOs). The protocol involves a straightforward mixing of reagents followed by a controlled heating period, after which standard workup procedures yield the pure product. The detailed standardized synthesis steps below outline the precise quantities and conditions required to replicate the high yields reported in the patent data, ensuring reproducibility from gram to kilogram scales.
- Mix palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and a nitro compound in an organic solvent like dioxane.
- Heat the reaction mixture to 120°C and stir for 16 to 30 hours to allow for carbonylation and cyclization.
- Filter the mixture, mix with silica gel, and purify via column chromatography to isolate the final 2-trifluoromethyl substituted quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this novel synthetic route offers transformative advantages that directly address the pain points of sourcing complex heterocyclic building blocks. The shift from expensive, pre-activated halides or high-pressure gas reagents to commodity chemicals like nitro compounds fundamentally alters the cost structure of the manufacturing process. Nitro compounds are ubiquitous in the chemical industry, produced in massive volumes for dyes and agrochemicals, which ensures a stable and competitive supply base. This abundance translates into significant cost savings and reduces the risk of supply disruptions that often plague specialized fine chemical markets. Moreover, the elimination of high-pressure carbon monoxide gas removes the need for specialized autoclaves and rigorous gas handling safety protocols, allowing production to occur in standard glass-lined reactors found in most multipurpose chemical plants. This flexibility enhances supply chain reliability by enabling production across a wider network of qualified manufacturers, reducing lead times and increasing the resilience of the global supply chain for these critical intermediates.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the substitution of costly reagents with inexpensive, commercially available starting materials. By utilizing nitro compounds and avoiding the need for pre-synthesized benzamides or anilines, the raw material costs are drastically simplified. Additionally, the use of solid Mo(CO)6 as a CO source eliminates the capital expenditure associated with high-pressure gas infrastructure and the ongoing operational costs of gas monitoring and safety compliance. The high reaction efficiency and yields reported, often exceeding 90% for optimized substrates, mean that less raw material is wasted, and the throughput per batch is maximized. This combination of lower input costs and higher output efficiency results in a substantially reduced cost of goods, providing a competitive edge in the pricing of final API intermediates.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals significantly de-risks the supply chain. Nitro compounds and simple imidoyl chlorides are produced by numerous suppliers globally, preventing single-source bottlenecks. The robustness of the reaction conditions, which tolerate a wide variety of functional groups, means that the process is less sensitive to minor variations in raw material quality, further stabilizing production schedules. The one-pot nature of the reaction reduces the number of unit operations, isolation steps, and solvent exchanges required, which shortens the overall manufacturing cycle time. This streamlined workflow allows for faster turnaround times from order to delivery, enabling pharmaceutical companies to accelerate their drug development timelines and respond more agilely to market demands without compromising on quality or regulatory compliance.
- Scalability and Environmental Compliance: Scaling this process from laboratory to industrial production is facilitated by the absence of hazardous high-pressure gases and the use of standard organic solvents like dioxane or acetonitrile. The reaction generates minimal waste compared to multi-step alternatives, as the atom economy is improved by the direct incorporation of the nitro group and the carbonyl source. The simplified post-treatment, involving filtration and standard chromatography or crystallization, reduces the volume of solvent waste and the energy consumption associated with extensive purification. This aligns with modern green chemistry principles and increasingly stringent environmental regulations, making it easier for manufacturers to obtain the necessary permits and maintain sustainable operations. The ability to scale safely and sustainably ensures long-term supply continuity for partners seeking reliable sources of complex fluorinated heterocycles.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent literature, ensuring accuracy and relevance for technical decision-makers evaluating this route for their own production needs.
Q: What are the key advantages of using nitro compounds in this synthesis?
A: Nitro compounds serve as inexpensive and readily available starting materials that replace the need for pre-activated amines or harsh reduction steps, significantly lowering raw material costs and simplifying the supply chain.
Q: How does this method improve safety compared to traditional carbonylation?
A: By utilizing solid Mo(CO)6 as a carbon monoxide substitute instead of high-pressure CO gas, the process eliminates the severe safety hazards associated with handling toxic gases under pressure, making it safer for industrial scale-up.
Q: What is the substrate scope for this reaction?
A: The method demonstrates excellent compatibility with various functional groups, including halogens, alkyls, and trifluoromethyl groups on both the aryl ring and the nitrogen substituent, allowing for the synthesis of diverse drug-like scaffolds.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes like the one described in CN112480015A for the development of next-generation therapeutics. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to industrial manufacturing is seamless and robust. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to handle fluorinated chemistry and transition metal catalysis safely and efficiently positions us as an ideal partner for bringing complex quinazolinone-based drugs to market.
We invite you to collaborate with us to leverage this advanced technology for your specific drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your project's unique requirements, demonstrating how this novel route can optimize your budget. Please contact us today to request specific COA data for related compounds and comprehensive route feasibility assessments, and let us help you secure a reliable supply of high-quality 2-trifluoromethyl quinazolinone intermediates for your global operations.