Advanced FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-Up
Advanced FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to access bioactive heterocyclic scaffolds, particularly those containing fluorine motifs which are critical for metabolic stability and bioavailability. A groundbreaking preparation method disclosed in patent CN111675662B introduces a highly efficient route for synthesizing 2-trifluoromethyl substituted quinazolinone compounds. This technology leverages a cost-effective iron-catalyzed cyclization strategy that transforms readily available trifluoroethylimidoyl chloride and isatin derivatives into valuable quinazolinone cores. For R&D directors and procurement managers alike, this represents a significant shift away from traditional, expensive synthons towards a more sustainable and economically viable manufacturing process. The ability to introduce the trifluoromethyl group directly during the ring-closing step simplifies the synthetic tree, reducing the overall step count and potentially lowering the cost of goods sold (COGS) for downstream API production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of quinazolinones bearing trifluoromethyl functional groups has relied heavily on the cyclization of pre-functionalized synthons such as anthranilamide, anthranilic acid, or isatoic anhydride with specific trifluoromethylating agents. Literature precedents often cite the use of trifluoroacetic anhydride or ethyl trifluoroacetate as the primary sources of the CF3 group. However, these conventional methodologies are frequently plagued by significant drawbacks that hinder their utility in large-scale commercial manufacturing. The reaction conditions are often severe, requiring harsh reagents that pose safety risks and complicate waste management. Furthermore, the starting materials, particularly specialized trifluoromethyl synthons, can be prohibitively expensive and difficult to source in bulk quantities. Narrow substrate scope is another critical limitation; many traditional methods fail to tolerate diverse functional groups, restricting the chemical space accessible to medicinal chemists and forcing them into lengthy protection-deprotection sequences that erode overall yield and efficiency.
The Novel Approach
In stark contrast to these legacy methods, the novel approach detailed in the patent utilizes a convergent strategy involving the direct coupling of trifluoroethylimidoyl chloride and isatin derivatives. This methodology capitalizes on the inherent reactivity of the imidoyl chloride moiety to form carbon-nitrogen bonds under mild alkaline promotion, followed by an elegant iron-catalyzed decarbonylation and cyclization cascade. By employing ferric chloride (FeCl3) as the catalyst, the process bypasses the need for precious metal catalysts like palladium or rhodium, which are not only costly but also subject to strict regulatory limits regarding residual metals in pharmaceutical products. The reaction proceeds in common aprotic solvents such as DMF under air atmosphere, eliminating the need for rigorous inert gas handling which simplifies reactor setup and operation. This robust protocol demonstrates exceptional functional group tolerance, accommodating electron-donating and electron-withdrawing substituents alike, thereby offering a versatile platform for generating diverse libraries of quinazolinone analogs for drug discovery programs.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The mechanistic pathway of this transformation is a sophisticated interplay of nucleophilic substitution and transition metal catalysis that ensures high regioselectivity and yield. Initially, the reaction involves an alkali-promoted formation of carbon-nitrogen bonds between the nitrogen of the isatin and the electrophilic carbon of the trifluoroethylimidoyl chloride. This step generates a key trifluoroacetamidine intermediate, which serves as the precursor for the subsequent ring closure. The presence of sodium hydride acts as a strong base to deprotonate the isatin nitrogen, enhancing its nucleophilicity and driving the initial coupling forward efficiently. Following this, the iron catalyst plays a pivotal role in facilitating the decarbonylation of the isatin moiety. This decarbonylation step is crucial as it removes the carbonyl oxygen from the five-membered ring of the isatin, allowing for the rearrangement and formation of the six-membered quinazolinone core. The trifluoromethyl group remains intact throughout this process, positioned perfectly at the 2-position of the final heterocycle.
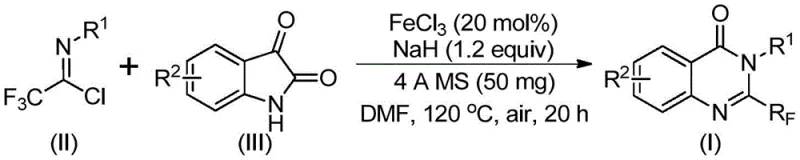
Following the decarbonylation, the system undergoes an isomerization and cyclization event to aromatize the ring, yielding the stable 2-trifluoromethyl substituted quinazolinone product. The use of 4A molecular sieves in the reaction mixture is a strategic choice to scavenge any trace moisture that could hydrolyze the sensitive imidoyl chloride starting material or deactivate the catalyst, thereby ensuring consistent high yields across different substrate combinations. The tolerance for various substituents (R1 and R2) suggests that the electronic nature of the aromatic rings does not significantly impede the catalytic cycle, likely due to the high activity of the iron species in promoting the C-N bond formation and subsequent rearrangement. This mechanistic robustness is what allows the process to achieve yields ranging significantly high across a broad panel of substrates, making it a reliable choice for synthesizing complex heterocyclic intermediates required in modern medicinal chemistry.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and thermal profiles to maximize conversion. The protocol dictates a two-stage heating process where the initial coupling occurs at a moderate temperature before ramping up to facilitate the cyclization. This controlled thermal gradient helps in managing the exothermicity of the initial bond formation while providing the necessary activation energy for the iron-catalyzed decarbonylation step. Detailed standard operating procedures regarding the addition order of sodium hydride and the handling of molecular sieves are critical for reproducibility. For a comprehensive guide on the exact molar ratios, solvent volumes, and workup procedures including column chromatography purification, please refer to the standardized synthesis steps outlined below.
- Combine ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in DMF solvent.
- Stir the reaction mixture at 40°C for approximately 10 hours to initiate the coupling, then increase temperature to 120°C.
- Maintain heating at 120°C for an additional 20 hours under air atmosphere, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this FeCl3-catalyzed route offers tangible strategic benefits that extend beyond mere technical feasibility. The shift from expensive, specialized trifluoromethylating agents to commodity chemicals like isatin and aromatic amines (used to make the imidoyl chloride) fundamentally alters the cost structure of the supply chain. Isatin is a widely produced bulk chemical with a stable global supply, reducing the risk of raw material shortages that often plague niche synthetic intermediates. Additionally, the replacement of precious metal catalysts with ferric chloride eliminates the volatility associated with the pricing of rare earth and noble metals, allowing for more accurate long-term budget forecasting and cost stabilization.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the drastic reduction in catalyst costs and the simplification of the purification workflow. Ferric chloride is orders of magnitude cheaper than palladium or copper catalysts typically used in cross-coupling reactions, leading to substantial savings in raw material expenditure. Furthermore, because iron is less toxic and easier to remove than many heavy metals, the downstream purification processes can be streamlined, potentially reducing the consumption of silica gel and solvents during chromatography. This efficiency translates directly into a lower cost per kilogram for the final API intermediate, enhancing the overall margin profile for the finished pharmaceutical product.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the use of commercially available starting materials that are not subject to complex geopolitical supply constraints. The precursors, such as substituted anilines and isatins, are produced by multiple vendors globally, ensuring a competitive market and preventing single-source bottlenecks. The robustness of the reaction conditions, which tolerate air and moisture better than many organometallic processes, also reduces the likelihood of batch failures due to environmental factors. This reliability ensures consistent delivery schedules and minimizes the need for safety stock, optimizing inventory turnover rates for the manufacturing facility.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this method aligns well with green chemistry principles by utilizing a base metal catalyst and avoiding hazardous reagents. The ability to run the reaction under air atmosphere simplifies the engineering requirements for large-scale reactors, as there is no need for extensive nitrogen purging systems. The waste stream generated is less hazardous compared to processes using heavy metals or aggressive fluorinating agents, simplifying waste treatment and disposal compliance. This ease of scale-up from gram to multi-kilogram levels ensures that the transition from clinical trial material to commercial production can be achieved rapidly without the need for extensive process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the operational parameters and expected outcomes. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing pipelines.
Q: What are the primary advantages of this FeCl3-catalyzed method over traditional trifluoroacetic anhydride routes?
A: This novel method utilizes readily available and inexpensive isatin and trifluoroethylimidoyl chloride as starting materials, avoiding the severe reaction conditions and expensive substrates often associated with trifluoroacetic anhydride or ethyl trifluoroacetate cyclizations. Furthermore, the use of cheap iron catalyst instead of precious metals significantly lowers the raw material costs.
Q: Does this synthesis protocol support a wide range of functional groups?
A: Yes, the patent data demonstrates excellent functional group tolerance, successfully synthesizing derivatives with methyl, fluoro, bromo, chloro, nitro, and methoxy substituents on both the aryl ring of the imidoyl chloride and the isatin core without significant loss in yield.
Q: Is this process suitable for large-scale industrial production?
A: The method is explicitly designed for scalability, utilizing simple operation steps, common organic solvents like DMF, and robust reaction conditions that have been expanded to gram-level synthesis, providing a strong foundation for industrial scale application.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the development of next-generation therapeutics. Our team of expert chemists has thoroughly analyzed the FeCl3-catalyzed cyclization technology and is fully prepared to leverage this methodology for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone intermediate meets the highest industry standards for quality and consistency.
We invite you to collaborate with us to optimize your supply chain and reduce your overall manufacturing costs. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to discuss your project specifics,索取 specific COA data, and obtain detailed route feasibility assessments that will demonstrate the clear commercial advantages of adopting this advanced synthesis platform for your pharmaceutical intermediates.