Advanced Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Pharmaceutical Manufacturing
The landscape of modern medicinal chemistry is increasingly defined by the incorporation of nitrogen-rich heterocycles, with the 1,2,4-triazole scaffold standing out as a privileged structure in drug design. As detailed in the groundbreaking patent CN113105402B, published in September 2022, a novel preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds has been established, addressing critical gaps in current synthetic methodologies. This innovation is particularly significant given the prevalence of this molecular framework in high-value therapeutics such as Maraviroc, Sitagliptin, and Deferasirox, as illustrated in the structural overview below. The ability to efficiently introduce both trifluoromethyl and acyl groups onto the triazole ring simultaneously represents a substantial leap forward, enhancing the physicochemical properties of potential drug candidates, including their metabolic stability and lipophilicity.
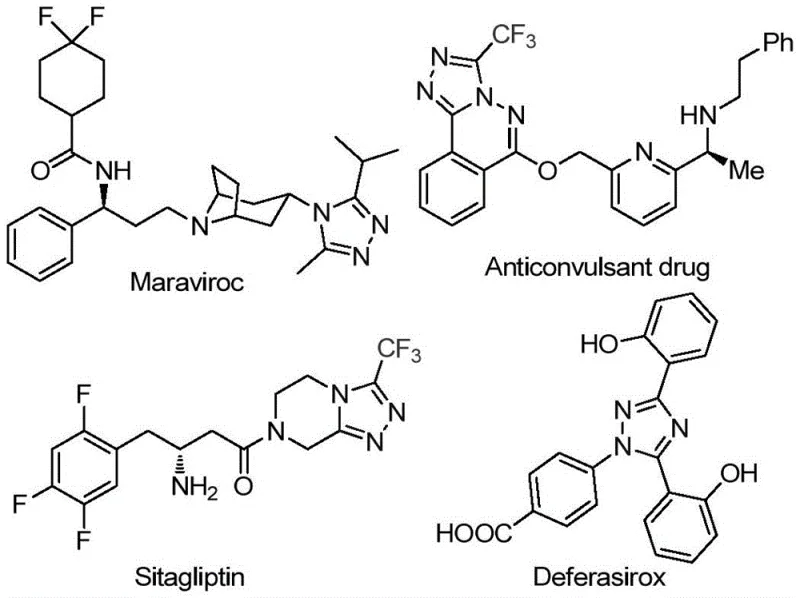
For R&D directors and process chemists, the introduction of fluorine atoms, specifically the trifluoromethyl group, into heterocyclic systems is often a bottleneck due to the harsh conditions typically required. The disclosed technology offers a streamlined pathway that bypasses these traditional hurdles. By leveraging a non-metallic iodine promotion system within a dimethyl sulfoxide (DMSO) solvent matrix, the method achieves high conversion rates while maintaining operational simplicity. This approach not only widens the applicability of triazole synthesis but also aligns perfectly with the industry's shift towards greener, more sustainable chemical manufacturing processes that minimize the use of toxic heavy metals.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles, especially those bearing specific functional groups like trifluoromethyl and acyl moieties, has relied heavily on transition metal catalysis or multi-step sequences involving sensitive intermediates. Conventional routes often necessitate stringent anhydrous and oxygen-free environments, requiring specialized equipment such as gloveboxes or Schlenk lines, which drastically increases capital expenditure and operational overhead. Furthermore, the reliance on precious metal catalysts like palladium or copper introduces significant challenges in downstream processing, as residual metal levels must be reduced to parts-per-million standards to meet regulatory requirements for pharmaceutical ingredients. These legacy methods frequently suffer from narrow substrate scope, poor atom economy, and the generation of hazardous waste streams, making them less attractive for cost-sensitive commercial manufacturing.
The Novel Approach
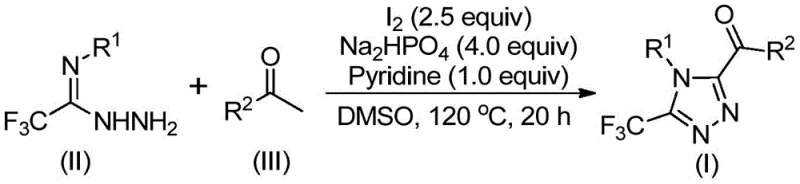
In stark contrast, the methodology described in patent CN113105402B utilizes a metal-free strategy driven by elemental iodine, a cheap and readily available reagent. This novel approach initiates with the iodination and Kornblum oxidation of aryl ethyl ketones in DMSO, generating reactive alpha-dicarbonyl intermediates in situ. These intermediates then undergo a tandem cyclization with trifluoroethylimide hydrazides under mild basic conditions facilitated by sodium dihydrogen phosphate and pyridine. The elimination of heavy metal catalysts not only reduces raw material costs but also simplifies the purification workflow, as there is no need for complex metal scavenging steps. The reaction tolerates a wide range of functional groups on both the aryl ketone and the hydrazide components, allowing for the rapid generation of diverse chemical libraries essential for lead optimization in drug discovery programs.
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
The core of this technological breakthrough lies in the synergistic interaction between elemental iodine and dimethyl sulfoxide, which acts as both the solvent and an oxidant. The mechanism likely proceeds through an initial alpha-iodination of the aryl ethyl ketone, followed by nucleophilic attack by DMSO to form a sulfonium intermediate. Subsequent hydrolysis or elimination yields the corresponding alpha-dicarbonyl species, which is the key electrophile for the subsequent ring closure. This in situ generation of the dicarbonyl precursor avoids the isolation of unstable intermediates, thereby improving overall process safety and yield. The trifluoroethylimide hydrazide then condenses with the dicarbonyl compound to form a hydrazone intermediate, which undergoes oxidative cyclization to forge the 1,2,4-triazole ring. The presence of pyridine and sodium dihydrogen phosphate serves to buffer the reaction medium, neutralizing the hydrogen iodide byproduct and driving the equilibrium towards the desired trisubstituted product.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based or high-temperature thermal cyclizations. The moderate reaction temperatures, ranging from 90°C to 130°C, minimize the risk of thermal decomposition of the sensitive trifluoromethyl group or the formation of polymeric byproducts. The use of DMSO ensures excellent solubility for both polar and non-polar substrates, creating a homogeneous reaction environment that promotes consistent kinetics and reduces the formation of side products associated with phase transfer issues. For quality assurance teams, this translates to a cleaner crude reaction profile, which significantly reduces the burden on final purification steps such as column chromatography or recrystallization, ultimately leading to higher purity specifications for the final active pharmaceutical ingredient (API) intermediate.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazoles Efficiently
The practical implementation of this synthesis route is designed for ease of adoption in both laboratory and pilot plant settings. The protocol involves a sequential addition strategy where the aryl ethyl ketone and iodine are first heated in DMSO to generate the oxidized intermediate, followed by the addition of the hydrazide and base components to trigger cyclization. This two-stage temperature profile allows for precise control over the reaction progression, ensuring complete conversion of the starting ketone before the introduction of the nitrogen source. Detailed standardized operating procedures for this specific transformation, including precise stoichiometric ratios and workup protocols, are outlined in the technical guide below to ensure reproducibility and safety during scale-up operations.
- Combine aryl ethyl ketone and elemental iodine in dimethyl sulfoxide (DMSO) and heat the mixture to 90-110°C for 4-6 hours to initiate oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the solution to 110-130°C for 12-20 hours to complete the cyclization, then filter and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iodine-promoted synthesis route presents a compelling value proposition centered on cost efficiency and supply reliability. The primary raw materials, aryl ethyl ketones and elemental iodine, are commodity chemicals available in bulk quantities from multiple global suppliers, mitigating the risk of single-source dependency. The elimination of expensive transition metal catalysts directly impacts the bill of materials, resulting in significant cost reduction in pharmaceutical intermediates manufacturing. Furthermore, the robustness of the reaction conditions means that the process is less susceptible to variations in raw material quality or minor fluctuations in environmental parameters, ensuring consistent batch-to-batch performance.
- Cost Reduction in Manufacturing: The most immediate financial benefit stems from the replacement of precious metal catalysts with inexpensive elemental iodine. In traditional cross-coupling or cyclization reactions, catalyst loading and subsequent removal can account for a substantial portion of the total production cost. By utilizing a metal-free system, manufacturers can eliminate the expense of metal scavengers and the associated waste disposal fees. Additionally, the use of DMSO, a low-cost polar aprotic solvent, further drives down operational expenditures compared to specialized fluorinated solvents often required for trifluoromethyl chemistry. The simplified workup procedure, which avoids complex extraction or filtration steps for metal removal, also reduces labor hours and utility consumption, contributing to a leaner and more profitable production model.
- Enhanced Supply Chain Reliability: Supply chain resilience is critically dependent on the availability and stability of key starting materials. The substrates used in this method, such as substituted acetophenones and trifluoroacetimidohydrazides, are widely produced and stocked by major chemical distributors worldwide. This widespread availability ensures that production schedules are not disrupted by raw material shortages. Moreover, the reaction does not require strictly anhydrous or anaerobic conditions, meaning that standard stainless steel reactors can be used without the need for specialized glass-lined vessels or inert gas blanketing systems. This flexibility allows for greater agility in scheduling production runs across different facilities, enhancing the overall continuity of supply for downstream customers.
- Scalability and Environmental Compliance: Scaling chemical processes from the bench to the tonne scale often reveals hidden bottlenecks related to heat transfer and exothermicity. The described method operates at moderate temperatures and utilizes a solvent with a high boiling point, providing a wide safety margin for large-scale operations. The absence of toxic heavy metals simplifies environmental compliance, as the effluent streams do not require rigorous treatment for metal contamination before discharge. This aligns with increasingly stringent global environmental regulations and corporate sustainability goals. The patent explicitly notes the ease of expanding the reaction to the gram level and beyond, indicating a clear path for commercial scale-up of complex pharmaceutical intermediates without the need for extensive process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel triazole synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a transparent view of the method's capabilities and limitations for potential partners and licensees.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the patented method utilizes elemental iodine as a non-metallic promoter, eliminating the need for costly palladium or copper catalysts and simplifying downstream purification.
Q: What are the advantages regarding reaction conditions?
A: The process operates effectively without strict anhydrous or oxygen-free conditions, significantly reducing operational complexity and equipment costs for large-scale production.
Q: Is this method suitable for industrial scale-up?
A: Yes, the patent explicitly states that the reaction can be easily expanded from gram-level laboratory synthesis to industrial scale production due to its robust and simple operational parameters.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient heterocycle synthesis in the development of next-generation therapeutics. Our team of expert process chemists has thoroughly evaluated the iodine-promoted pathway described in CN113105402B and confirmed its viability for large-scale production. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from clinical trials to market launch. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 3,4,5-trisubstituted 1,2,4-triazole intermediate meets the highest industry standards for identity and potency.
We invite you to collaborate with us to leverage this cost-effective and robust synthetic route for your specific drug development programs. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your volume requirements. We encourage you to contact us today to request specific COA data for our existing inventory or to discuss route feasibility assessments for your custom synthesis needs, ensuring a reliable supply of high-purity pharmaceutical intermediates for your global operations.