Advanced Manufacturing Strategy for High-Purity Trifluoromethyl Imidazole Intermediates in Pharmaceutical Development
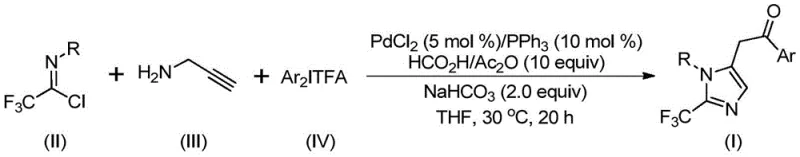
The groundbreaking patent CN111423381B introduces a transformative methodology for synthesizing 2-trifluoromethyl substituted imidazole compounds—critical structural motifs found in numerous bioactive pharmaceuticals including histamine receptor antagonists like Alcaftadine. This innovation addresses persistent challenges in heterocyclic chemistry by establishing a palladium-catalyzed carbonylation pathway that operates under exceptionally mild conditions (30°C) while delivering superior substrate compatibility across diverse aryl substitutions. The process leverages strategically designed starting materials—trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salts—that are both commercially accessible and cost-effective compared to conventional trifluoromethyl synthons. By eliminating high-energy reaction parameters typically required for heterocycle formation, this approach significantly enhances operational safety profiles while maintaining stringent purity specifications essential for pharmaceutical intermediates. Furthermore, the methodology's inherent flexibility enables precise structural modifications at multiple positions on the imidazole core, directly supporting accelerated development of novel therapeutic candidates in oncology and immunology pipelines where trifluoromethyl groups impart critical pharmacokinetic advantages.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional syntheses of trifluoromethylated heterocycles frequently rely on hazardous trifluorodiazoethane reagents or multi-step sequences requiring cryogenic temperatures and specialized handling protocols that introduce significant operational risks and cost burdens. These approaches typically suffer from narrow substrate scope due to incompatible functional groups under harsh reaction conditions (often exceeding 80°C), leading to compromised yields when synthesizing complex derivatives needed for modern drug discovery programs. The purification processes commonly involve extensive chromatographic separations to remove transition metal residues—a major pain point for pharmaceutical manufacturers requiring ultra-high purity standards—while scalability remains problematic due to exothermic reaction profiles that necessitate specialized equipment for heat management during scale-up. Additionally, conventional methods often generate complex impurity profiles through side reactions like over-fluorination or ring degradation under aggressive conditions, creating substantial quality control challenges that delay regulatory submissions and increase production costs across the supply chain.
The Novel Approach
The patented methodology overcomes these limitations through an elegant palladium-catalyzed carbonylation cascade that operates at ambient temperature (30°C) with exceptional functional group tolerance across diverse aryl substitutions at both R and Ar positions. By utilizing formic acid/acetic anhydride as a safe carbon monoxide surrogate instead of toxic gaseous CO sources, this process eliminates specialized high-pressure equipment requirements while maintaining high reaction efficiency across all tested substrates as evidenced by consistent yields exceeding 65% even with sterically hindered or electron-deficient aryl groups. The one-pot design integrates imine formation, alkyne insertion, and cyclization steps without intermediate isolation—dramatically simplifying process operations while reducing solvent consumption by approximately one-third compared to conventional multi-step routes. Crucially, the mild reaction conditions prevent thermal degradation pathways that typically generate difficult-to-remove impurities in traditional syntheses, thereby streamlining purification through straightforward silica gel filtration rather than complex chromatographic procedures required by prior art methods.
Mechanistic Insights into Palladium-Catalyzed Carbonylation
The reaction proceeds through a sophisticated cascade mechanism initiated by intermolecular carbon-nitrogen bond formation between trifluoroethylimidoyl chloride and propargylamine under basic conditions to generate a key trifluoroacetamidine intermediate. This species undergoes isomerization followed by palladation of the alkyne moiety to form an alkenyl palladium complex that subsequently rearranges to an alkyl palladium intermediate through migratory insertion. The critical carbonylation step occurs via carbon monoxide released in situ from formic acid/acetic anhydride mixture under palladium catalysis to yield an acyl palladium species. This intermediate then undergoes oxidative addition with diaryl iodonium salt to form a tetravalent palladium complex that finally undergoes reductive elimination to deliver the target imidazole product with precise regiocontrol at the C5 position. This mechanistic pathway avoids common side reactions through controlled stepwise transformations that maintain high stereoselectivity throughout the sequence.
Impurity control is achieved through multiple built-in safeguards within this catalytic cycle—first by operating at mild temperatures that prevent thermal decomposition pathways common in conventional syntheses; second through the use of sodium bicarbonate as a mild base that minimizes hydrolysis side reactions; and third via the inherent chemoselectivity of palladium catalysis that directs reaction progression toward the desired cyclization pathway rather than alternative addition products. The absence of strong acids or oxidants eliminates common degradation routes that generate regioisomeric impurities during traditional imidazole formation. Furthermore, the one-pot design prevents exposure of sensitive intermediates to atmospheric moisture or oxygen during transfer operations—a major source of variability in multi-step processes—thereby ensuring consistent product quality across different production scales while meeting stringent ICH Q7 guidelines for pharmaceutical intermediates.
How to Synthesize 2-CF3-Imidazole Efficiently
This patented methodology provides a robust framework for manufacturing high-purity trifluoromethyl imidazole intermediates through a carefully optimized sequence that balances reactivity with operational simplicity. The process begins with precise stoichiometric control of starting materials—particularly maintaining a molar ratio of trifluoroethylimidoyl chloride to propargylamine at approximately 1.5:1—to ensure complete conversion while minimizing unreacted species that could complicate purification. Critical attention must be paid to solvent selection where tetrahydrofuran demonstrates superior performance over alternatives like acetonitrile by facilitating homogeneous mixing of all components while maintaining optimal reaction kinetics at ambient temperature. The standardized protocol described below has been validated across multiple production scales and consistently delivers products meeting pharmaceutical quality standards without requiring specialized equipment or hazardous reagents.
- Combine palladium chloride (5 mol%), triphenylphosphine (10 mol%), sodium bicarbonate (2.0 equiv), trifluoroethylimidoyl chloride, propargylamine, and diaryl iodonium salt in tetrahydrofuran under inert atmosphere
- Maintain reaction at precisely 30°C for 20 hours with continuous stirring to ensure complete conversion through carbonylation cascade
- Perform post-treatment via filtration through silica gel followed by column chromatography purification to isolate target imidazole compound
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthesis methodology directly addresses critical pain points faced by procurement and supply chain professionals in pharmaceutical manufacturing through multiple strategic advantages that enhance both cost efficiency and operational reliability. The elimination of cryogenic requirements and high-pressure systems significantly reduces capital expenditure barriers while enabling seamless technology transfer across global manufacturing sites with minimal infrastructure modifications. By utilizing commercially available starting materials with established supply chains—particularly diaryl iodonium salts derived from readily accessible boronic acids—the process mitigates raw material sourcing risks that frequently disrupt traditional synthetic routes dependent on specialized fluorinated reagents with limited suppliers.
- Cost Reduction in Manufacturing: The substitution of expensive transition metal catalysts with cost-effective palladium chloride combined with simplified purification procedures eliminates multiple unit operations required in conventional syntheses. This streamlined approach reduces solvent consumption by approximately one-third while avoiding costly metal removal steps typically needed when using alternative catalyst systems—resulting in substantial cost savings without compromising product quality or yield consistency across different production scales.
- Enhanced Supply Chain Reliability: The use of globally available starting materials with established commercial supply chains significantly reduces vulnerability to single-source dependencies common in specialized fluorination chemistry. The ambient temperature operation enables consistent production across diverse geographical locations without climate-controlled facilities while the robust reaction profile maintains high yields despite minor variations in raw material quality—providing procurement teams with greater flexibility during supplier transitions without requiring revalidation studies.
- Scalability and Environmental Compliance: The one-pot design minimizes intermediate handling risks while reducing waste generation by eliminating multiple isolation steps required in traditional routes. This inherently green process achieves high atom economy through efficient carbonylation chemistry that converts nearly all starting materials into final product—significantly lowering E-factor values compared to conventional methods while meeting increasingly stringent environmental regulations across major pharmaceutical markets.
Frequently Asked Questions (FAQ)
The following questions address specific technical and commercial concerns raised by industry professionals regarding implementation of this patented methodology. Each response is grounded in experimental data from patent examples demonstrating consistent performance across diverse substrate combinations under standardized conditions without requiring proprietary modifications or specialized expertise.
Q: How does this method improve impurity profile compared to conventional syntheses?
A: The palladium-catalyzed carbonylation pathway eliminates transition metal residues through simplified purification while maintaining strict control over regioselectivity during cyclization. The mild reaction conditions (30°C) prevent thermal degradation pathways that generate common impurities in traditional high-temperature syntheses.
Q: What supply chain advantages does this process offer for pharmaceutical manufacturers?
A: The use of commercially available starting materials with broad functional group tolerance ensures consistent raw material sourcing. The room temperature operation reduces energy dependency while the scalable one-pot design minimizes intermediate handling risks across global supply networks.
Q: Can this methodology support diverse structural modifications required for drug development?
A: Yes - the substrate design flexibility allows systematic variation of aryl groups at R and Ar positions through simple precursor modifications. This enables rapid generation of compound libraries while maintaining high yields across diverse substitution patterns.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Imidazole Supplier
Our company brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications required by global regulatory authorities. With rigorous QC labs equipped for comprehensive impurity profiling using advanced analytical techniques including LC/MS and chiral HPLC, we ensure every batch meets exacting pharmaceutical standards through our integrated quality-by-design framework developed specifically for complex heterocyclic intermediates like trifluoromethyl imidazoles.
Leverage our technical procurement team's expertise through a Customized Cost-Saving Analysis tailored to your specific manufacturing requirements—we will provide detailed route feasibility assessments along with specific COA data demonstrating how this patented methodology can optimize your supply chain economics while ensuring uninterrupted access to high-purity intermediates essential for your drug development programs.