Advanced Iron-Catalyzed Cyclization for Scalable Production of Bioactive Quinazolinone Scaffolds
Advanced Iron-Catalyzed Cyclization for Scalable Production of Bioactive Quinazolinone Scaffolds
The pharmaceutical industry continuously seeks robust and cost-effective methodologies for constructing nitrogen-containing heterocycles, particularly quinazolinones, which serve as privileged scaffolds in medicinal chemistry. As highlighted in recent intellectual property developments, specifically patent CN111675662B, a significant breakthrough has been achieved in the efficient preparation of 2-trifluoromethyl substituted quinazolinone compounds. These structures are not merely academic curiosities; they are foundational cores for a wide array of bioactive molecules exhibiting potent anti-cancer, anticonvulsant, anti-inflammatory, and antifungal properties. The strategic introduction of the trifluoromethyl group further enhances these pharmacological profiles by improving metabolic stability, lipophilicity, and bioavailability, making them highly desirable targets for modern drug discovery programs.
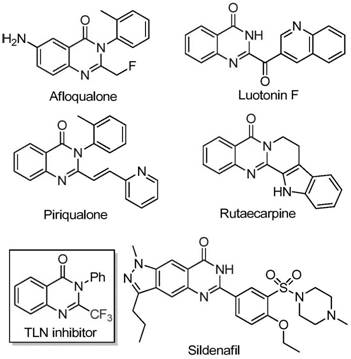
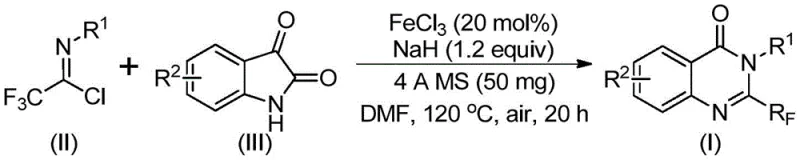
This novel synthetic approach addresses critical bottlenecks in the supply chain for high-purity pharmaceutical intermediates. By leveraging a transition metal-catalyzed cascade reaction, the process transforms readily available isatin derivatives and trifluoroethylimidoyl chlorides into complex fused ring systems with remarkable efficiency. For R&D directors and procurement managers alike, this represents a shift away from reliance on scarce resources towards a more sustainable and economically viable manufacturing paradigm. The ability to access these valuable scaffolds through a streamlined, iron-catalyzed pathway ensures a more reliable supply of key building blocks for next-generation therapeutics, reducing dependency on convoluted multi-step syntheses that often plague early-stage development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted quinazolinones has been fraught with significant chemical and economic challenges that hinder large-scale adoption. Traditional synthetic routes predominantly rely on the cyclization of trifluoromethyl-containing synthons, such as trifluoroacetic anhydride or ethyl trifluoroacetate, with substrates like anthranilamide or isatoic anhydride. While chemically feasible, these legacy methods suffer from severe limitations, including the requirement for harsh reaction conditions that can degrade sensitive functional groups, leading to lower overall yields and difficult purification processes. Furthermore, the starting materials often command high market prices due to complex upstream synthesis, and the narrow substrate scope restricts the ability of medicinal chemists to rapidly explore structure-activity relationships (SAR). These factors collectively contribute to inflated production costs and extended lead times, creating friction in the supply chain for potential API candidates.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN111675662B introduces a transformative strategy that utilizes cheap and easily obtainable starting materials to overcome these historical barriers. By employing trifluoroethylimidoyl chloride and isatin as the primary building blocks, the new route bypasses the need for expensive anhydrides and simplifies the molecular assembly process. The reaction is driven by an inexpensive iron catalyst, specifically ferric chloride, which facilitates a tandem decarbonylation and cyclization sequence under relatively mild thermal conditions. This approach not only broadens the functional group tolerance, allowing for the incorporation of diverse substituents like halogens and alkyl groups, but also significantly streamlines the operational workflow. The result is a versatile platform capable of generating a wide library of 2-trifluoromethyl quinazolinones, such as compounds I-1 through I-5, with high efficiency and minimal environmental footprint.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The success of this synthetic transformation lies in the intricate interplay between the base-promoted nucleophilic attack and the subsequent iron-catalyzed rearrangement. The mechanism initiates with the deprotonation of the isatin nitrogen by sodium hydride, generating a reactive nucleophile that attacks the electrophilic carbon of the trifluoroethylimidoyl chloride. This step forms a crucial carbon-nitrogen bond, yielding a trifluoroacetamidine intermediate. Unlike traditional pathways that might stall at this stage or require forcing conditions to proceed, the presence of ferric chloride acts as a Lewis acid to activate the carbonyl moiety for decarbonylation. This iron-mediated step is pivotal, as it triggers the intramolecular cyclization that closes the quinazolinone ring system, ultimately delivering the thermodynamically stable 2-trifluoromethyl product. The use of 4A molecular sieves further drives the equilibrium forward by sequestering moisture, ensuring high conversion rates even under aerobic conditions.
From an impurity control perspective, this mechanism offers distinct advantages for process chemistry teams aiming for high-purity outputs. The specificity of the iron catalyst minimizes side reactions such as polymerization or over-alkylation, which are common pitfalls in base-mediated heterocycle synthesis. The reaction conditions, involving a two-stage heating profile (initially at 40°C followed by 120°C), allow for controlled kinetics that favor the desired cyclization over competing degradation pathways. Moreover, the tolerance of the catalytic system towards various electronic environments on the aromatic rings means that electron-withdrawing or electron-donating groups do not significantly derail the reaction trajectory. This robustness ensures a clean crude reaction profile, simplifying downstream purification and reducing the burden on quality control laboratories to identify and quantify complex impurity spectra.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires adherence to specific operational parameters to maximize yield and safety. The protocol dictates a precise stoichiometric balance, typically utilizing a slight excess of the imidoyl chloride relative to the isatin substrate to ensure complete consumption of the limiting reagent. The reaction is conducted in polar aprotic solvents like DMF, which effectively solubilize both the organic substrates and the inorganic base. A critical aspect of the procedure is the thermal gradient; the mixture is first stirred at a moderate temperature of 40°C for approximately 8 to 10 hours to allow for the initial coupling, followed by heating to 120°C for an additional 18 to 20 hours to drive the cyclization to completion. Detailed standardized synthesis steps see the guide below.
- Prepare the reaction mixture by adding ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative into DMF solvent under air.
- Heat the mixture initially at 40°C for 8-10 hours to facilitate initial bond formation, then raise the temperature to 120°C and maintain for 18-20 hours to complete the cyclization.
- Upon completion, filter the reaction mixture, mix the residue with silica gel, and purify via column chromatography to isolate the high-purity 2-trifluoromethyl substituted quinazolinone product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this iron-catalyzed methodology translates into tangible strategic benefits that extend beyond simple chemical yield. The shift from precious metal catalysts to abundant iron salts fundamentally alters the cost structure of the manufacturing process, eliminating the volatility associated with rhodium or palladium markets. Additionally, the use of commodity chemicals like isatin and simple aromatic amines as precursors ensures a stable and diversified supply base, mitigating the risk of raw material shortages that can disrupt production schedules. The operational simplicity, characterized by air tolerance and straightforward workup procedures, reduces the need for specialized equipment and extensive operator training, thereby lowering the barrier to entry for contract manufacturing organizations (CMOs) looking to offer this capability.
- Cost Reduction in Manufacturing: The replacement of expensive noble metal catalysts with ferric chloride results in a drastic reduction in direct material costs, as iron is orders of magnitude cheaper and more accessible globally. Furthermore, the elimination of rigorous inert atmosphere requirements (such as gloveboxes or extensive nitrogen purging) lowers energy consumption and infrastructure overhead. The high atom economy of the cyclization step minimizes waste generation, which in turn reduces the costs associated with hazardous waste disposal and environmental compliance. These cumulative savings allow for a more competitive pricing model for the final pharmaceutical intermediate, enhancing margins for downstream API manufacturers.
- Enhanced Supply Chain Reliability: By utilizing starting materials that are widely produced for other industrial applications, such as isatin and various anilines, the supply chain becomes inherently more resilient to market fluctuations. The robustness of the reaction conditions means that production is less susceptible to minor variations in utility quality or environmental factors, ensuring consistent batch-to-batch quality. This reliability is crucial for maintaining continuous supply agreements with major pharmaceutical clients who demand strict adherence to delivery timelines. The ability to source catalysts and reagents from multiple global vendors further de-risks the procurement strategy, preventing single-source bottlenecks.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, having been demonstrated to proceed efficiently on gram scales with potential for tonnage production. The use of iron, a non-toxic and environmentally benign metal, aligns with green chemistry principles and simplifies regulatory filings regarding heavy metal residues in the final drug substance. The simplified post-treatment, involving basic filtration and chromatography, avoids complex extraction sequences that generate large volumes of organic waste. This eco-friendly profile not only meets increasingly stringent environmental regulations but also appeals to sustainability-focused stakeholders within the pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. Understanding these details is essential for R&D teams evaluating the feasibility of this chemistry for their specific pipeline projects. The answers provided are derived directly from the experimental data and beneficial effects reported in the patent literature, ensuring accuracy and relevance for decision-makers assessing this technology for potential licensing or process development.
Q: What are the primary advantages of using Ferric Chloride in this synthesis?
A: Ferric chloride serves as an inexpensive and earth-abundant catalyst compared to precious metals like palladium or rhodium. It effectively promotes the decarbonylation and cyclization steps while maintaining high functional group tolerance, significantly lowering the overall production cost.
Q: Can this method accommodate diverse substrate variations for drug discovery?
A: Yes, the protocol demonstrates excellent substrate scope. It tolerates various substituents on both the isatin ring (such as methyl, fluoro, bromo, and methoxy groups) and the imidoyl chloride aryl ring, allowing for the rapid generation of diverse libraries for SAR studies.
Q: Is this synthetic route suitable for industrial scale-up?
A: The method is designed for scalability. It utilizes readily available starting materials, operates under relatively mild conditions without stringent inert gas requirements (air tolerant), and employs a simple workup procedure, making it highly viable for commercial kilogram-to-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing high-quality heterocyclic building blocks to accelerate drug discovery timelines. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into robust manufacturing processes. We are committed to delivering 2-trifluoromethyl quinazolinones and related intermediates with stringent purity specifications, supported by our rigorous QC labs that employ state-of-the-art analytical instrumentation to verify every batch. Our dedication to quality assurance guarantees that the materials you receive meet the exacting standards required for preclinical and clinical development.
We invite you to collaborate with us to leverage this advanced iron-catalyzed technology for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline constraints. We encourage you to reach out today to obtain specific COA data for our catalog compounds or to discuss custom route feasibility assessments for novel analogs. Let us help you optimize your supply chain and reduce time-to-market with our reliable and cost-effective synthesis solutions.