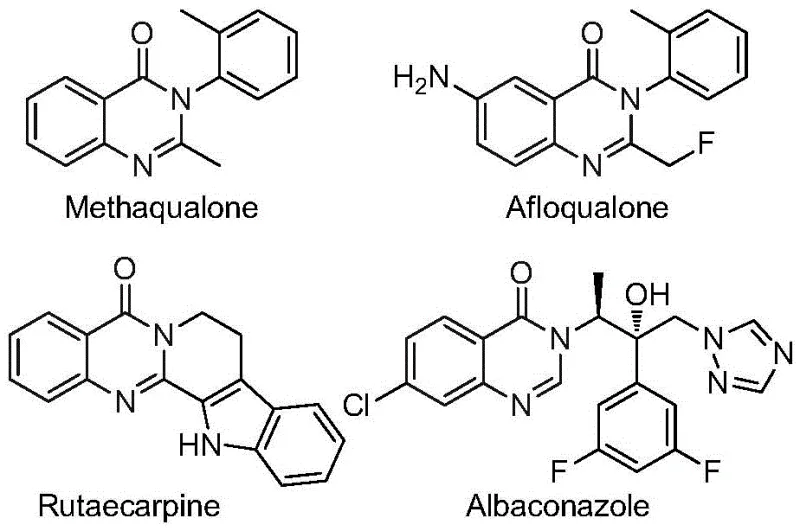
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Pharmaceutical Applications
The pharmaceutical industry continuously seeks efficient pathways to access privileged scaffolds that exhibit potent biological activity. Patent CN112480015B introduces a groundbreaking multi-component one-pot method for synthesizing 2-trifluoromethyl substituted quinazolinones, a core structure found in numerous bioactive molecules ranging from antifungals to anticancer agents. This technology leverages a sophisticated palladium-catalyzed carbonylation cascade that transforms inexpensive nitro compounds and trifluoroethylimidoyl chlorides into high-value heterocycles. By integrating a solid carbon monoxide source, Mo(CO)6, the process circumvents the safety hazards of high-pressure gas while maintaining exceptional reaction efficiency. For R&D teams focused on novel drug discovery, this methodology offers a versatile platform for generating diverse chemical libraries with improved metabolic stability conferred by the trifluoromethyl group.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core has been plagued by significant synthetic bottlenecks that hinder large-scale production and rapid iteration. Traditional routes often rely on the use of pre-activated substrates such as 2-bromoformylanilines or acid anhydrides, which are not only costly to procure but also require additional synthetic steps to prepare, thereby increasing the overall step count and waste generation. Furthermore, many established protocols necessitate the use of gaseous carbon monoxide under high-pressure conditions, demanding specialized autoclaves and rigorous safety protocols that are impractical for many standard laboratory or pilot plant settings. Other methods involving iron or ruthenium catalysis frequently suffer from narrow substrate scope, failing to tolerate sensitive functional groups, or require harsh reaction conditions that lead to poor yields and difficult purification processes. These limitations collectively inflate the cost of goods and extend the lead time for bringing new pharmaceutical candidates to market.
The Novel Approach
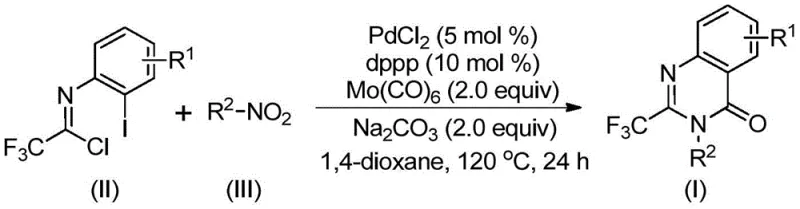
The methodology disclosed in CN112480015B represents a paradigm shift by utilizing readily available nitro compounds as the nitrogen source, effectively bypassing the need for expensive pre-functionalized anilines. This innovative route employs a palladium catalyst system in conjunction with Molybdenum Hexacarbonyl, which serves as a convenient solid surrogate for carbon monoxide, releasing the gas in situ under thermal conditions. This strategic modification allows the reaction to proceed at atmospheric pressure in standard glassware, drastically reducing capital expenditure on safety equipment. The one-pot nature of the reaction combines reduction, coupling, and cyclization events into a single operational sequence, minimizing intermediate isolation and solvent consumption. As illustrated in the reaction scheme below, this streamlined approach not only simplifies the workflow but also enhances the overall atom economy, making it an ideal candidate for cost reduction in API manufacturing.
Mechanistic Insights into Palladium-Catalyzed Carbonylation Cascade
The success of this transformation relies on a meticulously orchestrated catalytic cycle initiated by the reduction of the nitro group. Initially, the Molybdenum Hexacarbonyl acts as a reducing agent, converting the nitro compound into the corresponding amine intermediate under the reaction conditions. This freshly generated amine then undergoes a base-promoted nucleophilic attack on the trifluoroethylimidoyl chloride, forming a trifluoroacetamidine derivative in situ. Concurrently, the palladium catalyst, coordinated by the dppp ligand, inserts into the carbon-iodine bond of the imidoyl chloride moiety to generate a reactive organopalladium species. As the temperature reaches 120 °C, the molybdenum complex decomposes further to release carbon monoxide, which subsequently inserts into the carbon-palladium bond to form an acyl-palladium intermediate. This key acyl species then undergoes intramolecular cyclization facilitated by the base, closing the ring to form the quinazolinone skeleton before final reductive elimination releases the product and regenerates the active catalyst.
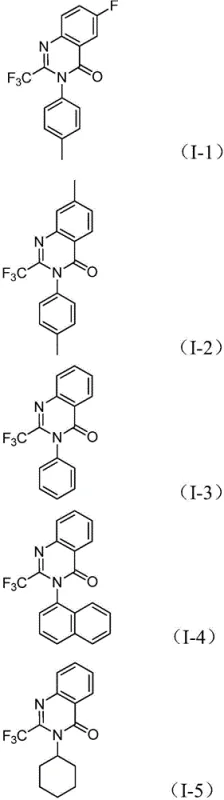
From an impurity control perspective, this mechanism offers distinct advantages over stepwise syntheses. By keeping the reactive amine and imidoyl chloride in the same pot, the concentration of free amine is kept low, minimizing the formation of bis-acylated side products that often plague amidation reactions. Furthermore, the use of a specific ligand system (dppp) stabilizes the palladium center, preventing the formation of palladium black and ensuring consistent catalytic turnover throughout the 16 to 30-hour reaction window. The tolerance for various substituents, as evidenced by the successful synthesis of derivatives bearing fluorine, chlorine, and methyl groups, indicates that the electronic properties of the substrate do not significantly inhibit the oxidative addition or CO insertion steps. This robustness ensures a clean impurity profile, which is critical for meeting the stringent purity specifications required for pharmaceutical intermediates.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
To implement this synthesis effectively, precise control over stoichiometry and reaction parameters is essential to maximize yield and minimize byproduct formation. The patent outlines a standardized protocol where the molar ratio of trifluoroethylimidoyl chloride to nitro compound is optimized at approximately 1:1.2, ensuring the nitro compound is in slight excess to drive the reduction-coupling sequence to completion. The catalyst loading is kept economical at 5 mol % PdCl2 with 10 mol % dppp ligand, balancing cost with reaction rate. Detailed standardized synthesis steps see the guide below.
- Combine palladium chloride (5 mol %), dppp ligand (10 mol %), sodium carbonate (2.0 equiv), Mo(CO)6 (2.0 equiv), trifluoroethylimidoyl chloride, and the specific nitro compound substrate in 1,4-dioxane solvent within a reaction vessel.
- Heat the reaction mixture to 120 °C and maintain stirring for a duration of 16 to 30 hours to ensure complete conversion of the starting materials into the desired heterocyclic product.
- Upon completion, filter the reaction mixture, mix the residue with silica gel, and purify the crude product via column chromatography to isolate the high-purity 2-trifluoromethyl substituted quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits that extend beyond simple yield metrics. The primary driver for cost optimization lies in the raw material selection; by shifting from expensive, pre-activated bromo-anilines to commodity-grade nitro compounds, the direct material cost is significantly reduced. Additionally, the elimination of high-pressure carbon monoxide cylinders removes the need for specialized gas handling infrastructure and associated safety audits, leading to substantial overhead savings in facility operations. The simplified one-pot procedure reduces solvent usage and labor hours associated with intermediate workups, further driving down the operational expenditure per kilogram of product. These factors combine to create a highly competitive cost structure for the manufacturing of these valuable heterocyclic intermediates.
- Cost Reduction in Manufacturing: The economic viability of this process is anchored in the use of earth-abundant starting materials and the avoidance of precious metal scavenging steps often required with other catalyst systems. By utilizing a solid CO source, the process eliminates the logistics costs and safety premiums associated with transporting and storing toxic gases. The high conversion rates observed across diverse substrates mean that less raw material is wasted in purification, directly improving the mass balance and reducing the cost of waste disposal. Consequently, this method enables a leaner manufacturing model that is resilient to fluctuations in the pricing of specialized reagents.
- Enhanced Supply Chain Reliability: Sourcing reliability is markedly improved because nitro compounds and trifluoroethylimidoyl chlorides are widely produced bulk chemicals with stable global supply chains, unlike niche heterocyclic building blocks that may have single-source suppliers. The robustness of the reaction conditions, which tolerate a wide range of functional groups, means that supply disruptions for specific substituted anilines can be mitigated by switching to alternative nitro precursors without re-validating the entire process. This flexibility ensures continuous production capability and reduces the risk of stockouts for critical API intermediates, safeguarding the downstream drug development timeline.
- Scalability and Environmental Compliance: The transition from laboratory to commercial scale is facilitated by the absence of high-pressure equipment, allowing the reaction to be run in standard stainless steel reactors equipped with standard heating jackets. The use of 1,4-dioxane as a solvent, while requiring careful recovery, is a well-established industrial solvent with existing recycling infrastructure. Furthermore, the high atom economy of the multi-component reaction generates less chemical waste per unit of product compared to linear synthesis routes, aligning with modern green chemistry principles and simplifying regulatory compliance regarding effluent discharge. This scalability ensures that the process can seamlessly grow from gram-scale R&D to multi-ton commercial production without fundamental changes to the unit operations.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this patented synthesis method. These insights are derived directly from the experimental data and mechanistic understanding provided in the patent documentation, offering clarity on reaction scope and operational parameters. Understanding these nuances is vital for process chemists aiming to adapt this technology for specific project needs.
Q: What is the primary advantage of using nitro compounds over traditional substrates in this synthesis?
A: Nitro compounds are significantly cheaper and more readily available than pre-activated substrates like 2-bromoformylanilines. This method eliminates the need for expensive starting materials and harsh pre-activation steps, streamlining the supply chain for API intermediates.
Q: How does the patent address the safety concerns associated with carbon monoxide usage?
A: The process utilizes Molybdenum Hexacarbonyl (Mo(CO)6) as a solid carbon monoxide substitute. This allows for the in-situ generation of CO under heating conditions, avoiding the logistical hazards and specialized high-pressure equipment required for handling gaseous carbon monoxide directly.
Q: Is this synthetic route compatible with diverse functional groups for drug discovery?
A: Yes, the method exhibits excellent substrate compatibility. It successfully tolerates various substituents including halogens (F, Cl, Br), alkyl groups, and trifluoromethyl groups on both the nitro compound and the imidoyl chloride, allowing for the rapid generation of diverse chemical libraries.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient synthetic methodologies play in accelerating drug development timelines. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to industrial manufacturing is seamless. We are committed to delivering high-purity 2-trifluoromethyl quinazolinones that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our dedication to quality assurance guarantees that every batch supplied adheres to the highest international standards, providing our partners with the confidence needed to advance their clinical programs.
We invite you to collaborate with us to leverage this advanced technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your supply chain and reduce overall project costs.