Advanced FeCl3-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Production
Introduction to Next-Generation Quinazolinone Manufacturing
The pharmaceutical industry continuously seeks robust synthetic methodologies for nitrogen-containing heterocycles, particularly quinazolinones, due to their pervasive presence in bioactive molecules ranging from anticancer agents to antifungal drugs. As highlighted in recent intellectual property developments, specifically patent CN111675662B, a significant breakthrough has been achieved in the efficient preparation of 2-trifluoromethyl substituted quinazolinone compounds. This class of molecules is highly prized because the introduction of a trifluoromethyl group significantly enhances metabolic stability, lipophilicity, and bioavailability, addressing key pharmacokinetic challenges in modern drug design. The disclosed technology moves beyond traditional limitations by utilizing a cost-effective iron-catalyzed system that operates under relatively mild conditions compared to historical precedents. For R&D directors and procurement specialists, this represents a pivotal shift towards more sustainable and economically viable manufacturing processes for high-value pharmaceutical intermediates.
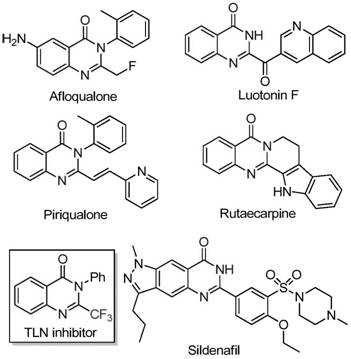
The strategic importance of this synthesis lies in its ability to access complex molecular scaffolds that were previously difficult or expensive to produce. By leveraging readily available starting materials such as isatin derivatives and trifluoroethylimidoyl chloride, the process circumvents the need for specialized, high-cost synthons often required in trifluoromethylation chemistry. This accessibility is crucial for supply chain heads who must ensure continuity of supply for critical API intermediates. Furthermore, the methodology described in the patent offers a wide substrate scope, allowing for the rapid generation of diverse analog libraries essential for lead optimization campaigns in medicinal chemistry. The integration of this technology into commercial production workflows promises to streamline the synthesis of key drug candidates, reducing both the environmental footprint and the overall cost of goods sold (COGS) for downstream pharmaceutical products.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted quinazolinones has been plagued by significant operational and economic hurdles that hinder large-scale adoption. Traditional routes often rely on the cyclization of anthranilamide or isatoic anhydride with expensive trifluoromethyl synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate. These conventional methods frequently suffer from severe reaction conditions, requiring harsh reagents that pose safety risks and generate substantial hazardous waste. Moreover, the substrate scope in these legacy processes is often narrow, limiting the structural diversity accessible to chemists during the drug discovery phase. Low yields and poor functional group tolerance further exacerbate the problem, necessitating complex purification steps that erode profit margins and extend production timelines. For procurement managers, these inefficiencies translate into volatile pricing and unreliable supply chains for critical intermediates, creating bottlenecks in the manufacturing of final active pharmaceutical ingredients.
The Novel Approach
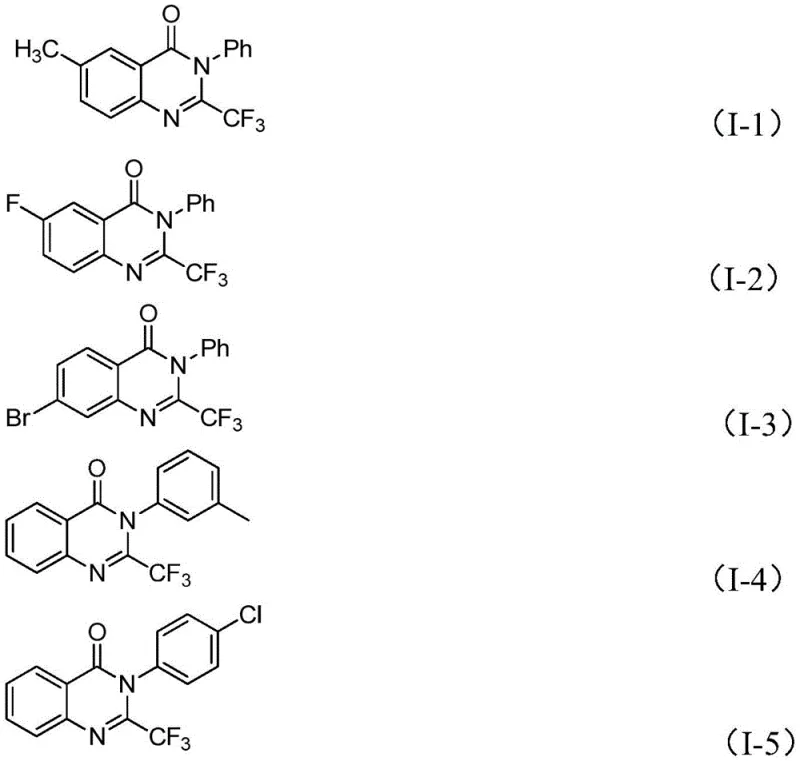
In stark contrast, the novel approach detailed in the patent utilizes a synergistic combination of ferric chloride and sodium hydride to catalyze the coupling of trifluoroethylimidoyl chloride with isatin derivatives. This method fundamentally alters the economic landscape of quinazolinone production by replacing precious metal catalysts with abundant and inexpensive iron salts. The reaction proceeds through a unique mechanism involving alkali-promoted carbon-nitrogen bond formation followed by iron-catalyzed decarbonylation and cyclization. This pathway not only improves atom economy but also operates under air atmosphere, eliminating the need for rigorous inert gas handling which simplifies reactor operations. The versatility of this approach is demonstrated by its compatibility with a wide array of substituents, including halogens and alkyl groups, allowing for the synthesis of diverse derivatives like compounds (I-1) through (I-5) with high efficiency. This technological leap provides a reliable foundation for the commercial scale-up of complex pharmaceutical intermediates, ensuring consistent quality and availability.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this innovative synthesis lies in the intricate interplay between the iron catalyst and the reaction substrates, which facilitates a transformative cyclization process. The mechanism initiates with the deprotonation of the isatin nitrogen by sodium hydride, generating a nucleophilic species that attacks the electrophilic carbon of the trifluoroethylimidoyl chloride. This step forms a key trifluoroacetamidine intermediate, setting the stage for the subsequent ring closure. The presence of ferric chloride is critical at this juncture, as it coordinates with the intermediate to promote decarbonylation, a step that is energetically demanding under standard conditions. The iron center likely stabilizes the transition state, lowering the activation energy barrier and driving the reaction towards the formation of the quinazolinone core. Understanding this mechanistic pathway is vital for R&D teams aiming to optimize reaction parameters or adapt the chemistry to novel substrates, as it highlights the specific roles of each reagent in the catalytic cycle.
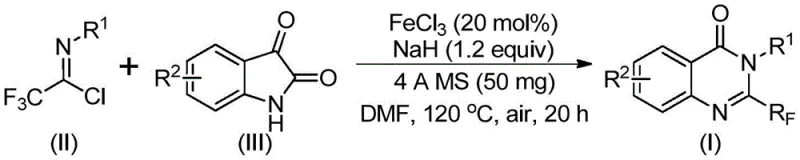
Furthermore, the inclusion of 4A molecular sieves plays a subtle yet profound role in controlling the impurity profile of the final product. By sequestering trace amounts of water generated during the reaction or present in the solvent, the molecular sieves prevent the hydrolysis of the sensitive imidoyl chloride starting material and the intermediate species. This moisture control is essential for maintaining high yields and minimizing the formation of side products such as carboxylic acids or amides, which can be difficult to separate from the target quinazolinone. The reaction conditions, specifically the two-stage heating profile starting at 40°C and ramping to 120°C, are finely tuned to balance the kinetics of intermediate formation with the thermodynamics of the final cyclization. This precise control over the reaction environment ensures that the process is not only chemically efficient but also robust enough for transfer to pilot and production scales, delivering high-purity intermediates suitable for stringent regulatory requirements.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
The practical implementation of this synthesis requires careful attention to reagent stoichiometry and thermal management to maximize yield and purity. The protocol outlined in the patent provides a clear roadmap for executing this transformation, emphasizing the importance of using anhydrous conditions and specific molar ratios to drive the reaction to completion. Operators must ensure that the ferric chloride catalyst is fully dispersed and that the sodium hydride is handled with appropriate safety precautions due to its pyrophoric nature. The use of dimethylformamide (DMF) as the solvent is preferred due to its ability to dissolve both organic substrates and inorganic salts, facilitating homogeneous reaction conditions. Detailed standard operating procedures for this synthesis, including specific workup and purification steps, are essential for reproducibility and are summarized in the technical guide below.
- Combine ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in anhydrous DMF solvent within a Schlenk tube.
- Initiate the reaction by stirring the mixture at 40°C for 8 to 10 hours to facilitate the initial nucleophilic attack and bond formation.
- Elevate the temperature to 120°C and maintain heating for 18 to 20 hours under air atmosphere to drive the decarbonylation and cyclization to completion, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this iron-catalyzed methodology offers compelling strategic advantages that extend beyond simple chemical efficiency. The primary benefit is the drastic reduction in raw material costs associated with the catalyst system. By substituting expensive palladium or rhodium complexes with commodity-grade ferric chloride, manufacturers can achieve significant cost savings per kilogram of produced intermediate. This cost structure improvement is compounded by the use of readily available starting materials like isatin and aromatic amines, which are produced on a multi-ton scale globally, ensuring a stable and competitive supply base. The elimination of precious metals also simplifies the downstream purification process, as there is no need for specialized scavenging resins or extensive testing for heavy metal residues, thereby reducing processing time and waste disposal costs.
- Cost Reduction in Manufacturing: The economic impact of switching to an iron-catalyzed system is profound, as it removes the volatility associated with precious metal markets from the cost equation. Ferric chloride is not only inexpensive but also used in low catalytic loadings (20 mol%), further minimizing material expenses. Additionally, the reaction operates under air atmosphere, removing the capital and operational expenditures related to maintaining inert nitrogen or argon environments in large reactors. These factors collectively contribute to a leaner manufacturing process with a lower break-even point, allowing for more competitive pricing of the final API. The simplified workup procedure, involving basic filtration and chromatography, reduces solvent consumption and labor hours, driving further efficiencies in the production budget.
- Enhanced Supply Chain Reliability: Supply chain resilience is significantly bolstered by the reliance on commodity chemicals rather than specialized, single-source reagents. Isatin and its derivatives are widely manufactured by multiple global suppliers, mitigating the risk of supply disruptions that can occur with niche catalysts or complex synthons. The robustness of the reaction conditions, which tolerate a variety of functional groups and operate effectively in standard solvents like DMF, means that the process can be easily transferred between different manufacturing sites without extensive re-validation. This flexibility allows supply chain heads to diversify their vendor base and secure long-term contracts with favorable terms, ensuring uninterrupted production schedules for critical drug substances. The scalability of the process from gram to kilogram levels demonstrates its readiness for commercial deployment, providing confidence in future supply continuity.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this synthesis aligns well with green chemistry principles by utilizing a non-toxic iron catalyst and avoiding hazardous reagents often found in traditional trifluoromethylation protocols. The high atom economy and reduced waste generation simplify the management of effluent streams, lowering the costs associated with environmental compliance and waste treatment. The ability to run the reaction at elevated temperatures (120°C) without specialized high-pressure equipment facilitates straightforward scale-up in standard glass-lined or stainless steel reactors. This ease of scale-up ensures that production volumes can be rapidly increased to meet market demand without the need for significant capital investment in new infrastructure. Consequently, manufacturers can respond agilely to market fluctuations while maintaining a strong sustainability profile.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific applications, we have compiled answers to common inquiries regarding the reaction scope, purification, and scalability. These insights are derived directly from the experimental data and technical specifications provided in the patent literature, ensuring accuracy and relevance for process development scientists. Understanding these nuances is critical for making informed decisions about integrating this synthetic route into existing manufacturing pipelines. The following section addresses key technical considerations that often arise during the technology transfer and optimization phases.
Q: What are the primary advantages of using FeCl3 over precious metal catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) eliminates the need for expensive palladium or rhodium catalysts, drastically reducing raw material costs and simplifying the removal of heavy metal residues, which is critical for meeting stringent pharmaceutical purity specifications.
Q: Does this synthesis method tolerate diverse functional groups on the isatin substrate?
A: Yes, the protocol demonstrates excellent functional group tolerance, successfully accommodating substrates with methyl, fluoro, bromo, chloro, and methoxy substituents at various positions (ortho, meta, para) without compromising yield or requiring protective group strategies.
Q: How does the two-stage temperature profile (40°C then 120°C) impact the reaction outcome?
A: The initial lower temperature phase (40°C) allows for the controlled formation of the intermediate trifluoroacetamidine species, while the subsequent high-temperature phase (120°C) provides the necessary activation energy for the iron-catalyzed decarbonylation and final ring closure, ensuring high conversion rates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic methodologies like the FeCl3-catalyzed cyclization described in patent CN111675662B. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate such innovative laboratory protocols into robust, commercial-scale manufacturing processes. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from R&D to market supply. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of novel quinazolinone derivatives or reliable supply of known compounds like CAS 49579-40-0, our capabilities are aligned with your most demanding requirements.
We invite you to collaborate with us to leverage these cost-effective and scalable synthetic routes for your next pharmaceutical project. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific molecule, identifying opportunities to optimize your supply chain and reduce overall manufacturing costs. We encourage you to contact our technical procurement team today to request specific COA data, discuss route feasibility assessments, and explore how our expertise in iron-catalyzed transformations can accelerate your drug development timeline. Let us be your trusted partner in navigating the complexities of fine chemical manufacturing and bringing life-saving medicines to patients faster.