Advanced One-Pot Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Manufacturing
Introduction to Next-Generation Quinazolinone Synthesis
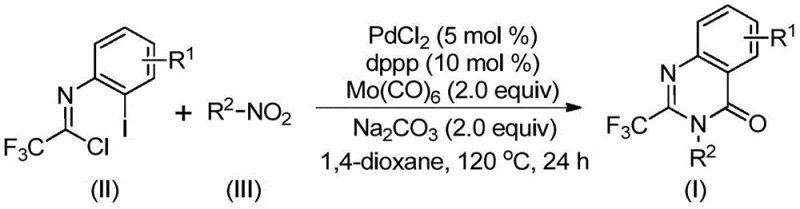
The quinazolinone scaffold represents a cornerstone structure in modern medicinal chemistry, underpinning a vast array of bioactive molecules with potent antifungal, antiviral, anti-inflammatory, and anticancer properties. Notable pharmaceutical agents such as methaqualone and various kinase inhibitors rely on this privileged heterocyclic framework. The introduction of a trifluoromethyl group at the 2-position further enhances these molecules by improving metabolic stability, lipophilicity, and bioavailability, making them highly desirable targets for drug discovery programs. Addressing the critical need for efficient access to these scaffolds, the technology disclosed in patent CN112480015B presents a groundbreaking multicomponent one-pot strategy. This innovation shifts the paradigm from multi-step, hazardous procedures to a streamlined, catalytic cascade that leverages inexpensive nitro compounds and trifluoroethylimidoyl chlorides.
For R&D directors and process chemists, the ability to construct complex nitrogen-containing heterocycles in a single operational step is a significant value driver. The disclosed method utilizes a sophisticated palladium-catalyzed carbonylation cascade that not only simplifies the synthetic route but also dramatically expands the accessible chemical space. By avoiding the isolation of unstable intermediates and utilizing a solid carbon monoxide source, this approach mitigates many of the safety and logistical bottlenecks associated with traditional heterocycle synthesis. As we delve deeper into the technical specifics, it becomes clear that this methodology offers a robust platform for the commercial scale-up of complex pharmaceutical intermediates, ensuring a reliable supply chain for high-value drug candidates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of quinazolinone rings has been fraught with synthetic challenges that hinder efficient manufacturing. Traditional pathways often rely on the condensation of anthranilic acid derivatives with amines or nitriles, which can suffer from poor atom economy and require harsh activation conditions. More advanced transition-metal catalyzed approaches have attempted to address these issues but frequently introduce new complications. For instance, ruthenium or platinum-catalyzed reductive N-heterocyclization typically necessitates the use of high-pressure carbon monoxide gas, demanding specialized autoclave reactors and rigorous safety protocols that increase capital expenditure. Furthermore, alternative iron-catalyzed condensations or palladium-mediated cyclizations involving 2-bromoformylaniline often require expensive, pre-functionalized substrates that are not readily available on a commodity scale.
These conventional limitations translate directly into higher production costs and longer lead times for high-purity pharmaceutical intermediates. The need for pre-activation of substrates adds extra synthetic steps, each introducing potential yield losses and purification burdens. Additionally, the narrow substrate scope of many older methods restricts the ability of medicinal chemists to rapidly explore structure-activity relationships (SAR). When a synthetic route cannot tolerate diverse functional groups or requires exotic reagents, it becomes a bottleneck in the drug development pipeline. Consequently, there is a persistent industry demand for a method that combines operational simplicity with broad chemical compatibility, eliminating the reliance on hazardous gases and costly starting materials.
The Novel Approach
The methodology described in patent CN112480015B offers a transformative solution by employing a palladium-catalyzed multicomponent coupling reaction that proceeds under relatively mild conditions. Central to this innovation is the use of molybdenum hexacarbonyl [Mo(CO)₆] as a safe, solid surrogate for carbon monoxide gas. This allows the carbonylation step to occur in standard reaction vessels at atmospheric pressure, removing the need for high-pressure infrastructure. The reaction seamlessly integrates the reduction of a nitro group, the formation of a carbon-nitrogen bond, and the subsequent cyclization into a single pot. By utilizing cheap and commercially available nitro compounds alongside trifluoroethylimidoyl chlorides, the process achieves high atom economy and significantly reduces raw material costs.
Furthermore, this novel approach exhibits exceptional functional group tolerance, accommodating a wide range of substituents on both the nitroarene and the imidoyl chloride components. The system effectively handles electron-withdrawing and electron-donating groups, as well as halogens, without compromising yield or selectivity. This versatility is crucial for the rapid generation of diverse libraries for biological screening. The operational simplicity is further enhanced by the use of common organic solvents like 1,4-dioxane and straightforward workup procedures involving filtration and column chromatography. For procurement managers, this translates to a reliable pharmaceutical intermediate supplier capability, where the supply chain is secured by the availability of bulk commodity chemicals rather than bespoke, expensive reagents.
Mechanistic Insights into Pd-Catalyzed Carbonylative Cyclization
The mechanistic pathway of this transformation is a testament to the elegance of modern organometallic catalysis, orchestrating multiple bond-forming events in a concerted sequence. The reaction initiates with the reduction of the nitro compound (III) to the corresponding amine by Mo(CO)₆, which serves a dual role as both the CO source and the reducing agent. This generated amine then undergoes a base-promoted nucleophilic attack on the trifluoroethylimidoyl chloride (II), forming a trifluoroacetamidine intermediate in situ. Concurrently, the palladium catalyst, generated from PdCl₂ and the dppp ligand, undergoes oxidative addition into the carbon-iodine bond of the imidoyl chloride moiety (or a related aryl iodide species depending on the specific substrate design implied in the broader scope), creating a reactive organopalladium(II) species.
As the temperature is maintained at 120°C, the Mo(CO)₆ thermally decomposes to release carbon monoxide, which subsequently inserts into the carbon-palladium bond to form an acyl-palladium intermediate. This key step introduces the carbonyl functionality essential for the quinazolinone ring closure. Under the influence of the base (sodium carbonate), an intramolecular nucleophilic attack by the nitrogen atom onto the acyl-palladium center occurs, leading to the formation of a seven-membered palladacycle. The catalytic cycle concludes with a reductive elimination step that releases the final 2-trifluoromethyl substituted quinazolinone product (I) and regenerates the active Pd(0) catalyst. This intricate dance of reduction, coupling, insertion, and cyclization ensures high efficiency and minimizes side reactions, providing a clean impurity profile that is vital for downstream processing.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and reaction parameters to maximize yield and purity. The protocol dictates a precise molar ratio of catalyst to ligand to base, typically utilizing 5 mol% PdCl₂ and 10 mol% dppp relative to the limiting reagent. The reaction is conducted in anhydrous 1,4-dioxane at 120°C for a duration of 16 to 30 hours, ensuring complete conversion of the starting materials. Detailed standardized operating procedures regarding the addition order of reagents, specifically the timing of Mo(CO)₆ addition to control CO release, are critical for reproducibility. For a comprehensive, step-by-step guide including exact quantities and workup details, please refer to the structured synthesis protocol below.
- Combine palladium chloride, dppp ligand, sodium carbonate, Mo(CO)6, trifluoroethylimidoyl chloride, and nitro compound in an organic solvent like 1,4-dioxane.
- Heat the reaction mixture to 120°C and maintain stirring for 16 to 30 hours to allow for nitro reduction, amidation, and cyclization.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target quinazolinone derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers compelling advantages that directly impact the bottom line and supply chain resilience. The primary driver for cost optimization lies in the selection of starting materials; nitro compounds are among the most abundant and inexpensive building blocks in the fine chemical industry, available in tonnage quantities from multiple global suppliers. By bypassing the need for pre-synthesized amines or activated carboxylic acid derivatives, the process eliminates several upstream manufacturing steps, thereby reducing the cumulative cost of goods sold (COGS). Additionally, the replacement of gaseous carbon monoxide with solid Mo(CO)₆ removes the requirement for specialized high-pressure reactors, allowing production to occur in standard glass-lined steel reactors found in most multipurpose chemical plants.
- Cost Reduction in Manufacturing: The economic benefits of this method are substantial due to the elimination of expensive catalysts like ruthenium or platinum in favor of more affordable palladium systems used at low loadings. The one-pot nature of the reaction significantly reduces solvent consumption and energy usage associated with intermediate isolations and drying steps. Furthermore, the high yields reported (often exceeding 90% for optimized substrates) minimize waste generation and maximize the throughput of the final API intermediate. This efficiency translates to a lower cost per kilogram of the active pharmaceutical ingredient, providing a competitive edge in pricing negotiations.
- Enhanced Supply Chain Reliability: Relying on commodity chemicals like nitrobenzenes and simple imidoyl chlorides ensures a robust and diversified supply base, reducing the risk of shortages associated with bespoke reagents. The robustness of the reaction conditions means that slight variations in raw material quality can be tolerated without catastrophic failure of the batch, enhancing process reliability. For supply chain heads, this means shorter lead times for high-purity pharmaceutical intermediates and the ability to scale production rapidly in response to market demand without waiting for complex custom synthesis of starting materials.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated effectively at the gram level with clear pathways to kilogram and ton-scale production. The use of a closed system for CO generation minimizes fugitive emissions, aligning with increasingly stringent environmental regulations regarding volatile organic compounds and toxic gases. The simplified workup, involving basic filtration and chromatography, reduces the volume of aqueous waste streams compared to traditional acid/base extraction methods. This green chemistry profile not only lowers waste disposal costs but also supports corporate sustainability goals, making the manufacturing process more attractive to environmentally conscious partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity on the practical application of the technology for industrial partners.
Q: What are the key advantages of using nitro compounds over pre-formed amines in this synthesis?
A: Using nitro compounds eliminates the need for separate amine synthesis steps, reducing overall process complexity and cost. Nitro compounds are widely available, inexpensive commodity chemicals with diverse substitution patterns, allowing for greater structural flexibility in the final API intermediate.
Q: How does the use of Mo(CO)6 improve safety compared to traditional carbonylation methods?
A: Traditional carbonylation often requires high-pressure carbon monoxide gas, which poses significant safety and infrastructure challenges. Mo(CO)6 acts as a solid, easy-to-handle CO surrogate that releases carbon monoxide in situ under heating, enabling the reaction to proceed in standard laboratory glassware without specialized high-pressure equipment.
Q: Is this synthetic route scalable for industrial production?
A: Yes, the patent explicitly demonstrates that the method can be expanded to the gram level and is suitable for industrial production. The use of robust catalysts, common solvents like dioxane, and the one-pot nature of the reaction simplify scale-up and post-treatment processes significantly.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes in accelerating drug development timelines. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries are seamlessly translated into viable manufacturing processes. We are equipped with state-of-the-art facilities capable of handling sensitive organometallic reactions under strict safety protocols, guaranteeing stringent purity specifications for every batch of 2-trifluoromethyl quinazolinone delivered. Our rigorous QC labs employ advanced analytical techniques to verify identity and purity, providing our clients with the confidence they need to move forward with clinical trials and regulatory filings.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and target specifications. We encourage potential partners to contact us directly to obtain specific COA data for our catalog compounds or to discuss route feasibility assessments for novel derivatives. Let us help you optimize your supply chain and reduce time-to-market with our reliable, high-quality pharmaceutical intermediate solutions.