Advanced Palladium-Catalyzed Synthesis of 3-Alkenyl Quinolin-2(1H)one Derivatives for Commercial Scale-Up
Advanced Palladium-Catalyzed Synthesis of 3-Alkenyl Quinolin-2(1H)one Derivatives for Commercial Scale-Up
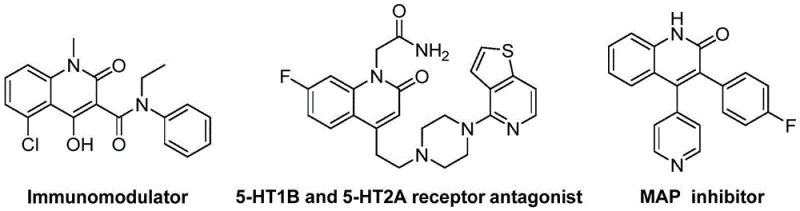
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable, and cost-effective methodologies for constructing nitrogen-containing heterocycles, which serve as the backbone for countless bioactive molecules. A groundbreaking development in this arena is detailed in patent CN114478375A, which discloses a highly efficient preparation method for 3-alkenyl quinolin-2(1H)one derivatives. These scaffolds are not merely academic curiosities; they are critical building blocks found in a vast array of therapeutic agents, ranging from antibiotics and antitumor drugs to antiplatelet agents and receptor antagonists. The significance of this technology lies in its ability to streamline the synthesis of these complex structures using readily available starting materials, thereby addressing key pain points for R&D Directors and Procurement Managers alike who are tasked with optimizing supply chains for high-purity pharmaceutical intermediates.
The biological relevance of the quinolin-2(1H)one core cannot be overstated, as evidenced by its presence in potent drugs such as nybomycin and deoxynybomycin. The versatility of this scaffold allows for extensive structural modification, enabling medicinal chemists to fine-tune pharmacokinetic properties. However, traditional synthetic routes often suffer from limitations such as harsh reaction conditions, poor atom economy, or the requirement for hazardous reagents. The novel approach presented in the patent data overcomes these hurdles by employing a palladium-catalyzed reductive aminocarbonylation strategy. This method leverages o-nitrobenzaldehyde as a dual-purpose reagent, acting simultaneously as the nitrogen source and the formyl source, which represents a significant departure from conventional multi-step syntheses that require separate introduction of these functionalities.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinolinone ring system has relied on classical condensation reactions or transition metal-catalyzed couplings that often demand stringent conditions. Traditional carbonylation reactions typically utilize aryl or vinyl halides as electrophiles, which can be expensive and generate stoichiometric amounts of salt waste. Furthermore, when allyl compounds are employed as electrophiles in carbonylation processes, the literature predominantly focuses on allyl chlorides, acetates, carbonates, or phosphates. These substrates, while effective, often present challenges regarding stability, toxicity, or availability. Allyl ethers, despite being natural, low-toxicity, and easy to handle, have received considerably less attention in carbonylation chemistry due to their lower reactivity as electrophiles. Consequently, developing a catalytic system capable of activating allyl aryl ethers for direct incorporation into the quinolinone framework has remained a formidable challenge for synthetic chemists, limiting the accessible chemical space for drug discovery programs.
The Novel Approach
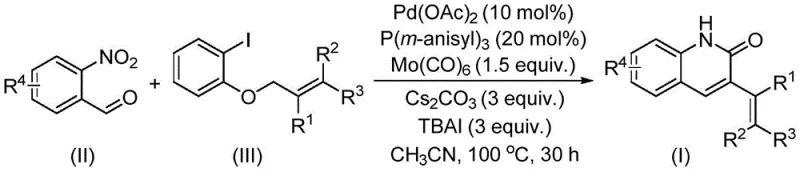
The methodology described in patent CN114478375A introduces a paradigm shift by successfully utilizing allyl aryl ethers as electrophiles in a palladium-catalyzed aminocarbonylation reaction. This innovative route reacts o-nitrobenzaldehyde with allyl aryl ethers in the presence of a specialized catalyst system comprising palladium acetate, tris(3-methoxyphenyl)phosphine, and molybdenum carbonyl. The reaction proceeds smoothly at 100 °C in acetonitrile, delivering the desired 3-alkenyl quinolin-2(1H)one derivatives in good to excellent yields. By bypassing the need for pre-functionalized allyl halides and utilizing the nitro group for in-situ reduction and cyclization, this process drastically simplifies the synthetic workflow. The ability to tolerate a wide range of functional groups on both the aldehyde and the ether components underscores the robustness of this method, making it an ideal candidate for the rapid generation of diverse compound libraries for biological screening.
Mechanistic Insights into Palladium-Catalyzed Reductive Aminocarbonylation
The success of this transformation hinges on the intricate interplay between the palladium catalyst and the carbonyl source provided by molybdenum carbonyl. The mechanism likely initiates with the oxidative addition of the palladium species to the allyl aryl ether, facilitated by the specific electronic properties of the tris(3-methoxyphenyl)phosphine ligand. This step generates a reactive pi-allyl palladium intermediate, which is crucial for the subsequent carbonylation event. The presence of molybdenum carbonyl serves as a solid, easy-to-handle source of carbon monoxide, which inserts into the palladium-carbon bond to form an acyl-palladium species. Simultaneously, the o-nitrobenzaldehyde undergoes a reductive process, potentially mediated by the metal carbonyl or the catalytic cycle itself, to generate the necessary amine functionality in situ. This amine then attacks the acyl-palladium intermediate, followed by cyclization and elimination to furnish the final quinolinone product. This cascade sequence effectively merges carbonylation, reduction, and cyclization into a single operational step, showcasing remarkable atom economy.
From an impurity control perspective, the choice of additives plays a pivotal role in ensuring high purity profiles essential for pharmaceutical applications. The inclusion of cesium carbonate as a base and tetrabutylammonium iodide as an additive helps to stabilize the catalytic cycle and suppress side reactions such as homocoupling or beta-hydride elimination. The reaction conditions are optimized to minimize the formation of regioisomers or over-reduced byproducts. For instance, maintaining the reaction temperature at 100 °C for 30 hours ensures complete conversion of the starting materials without degrading the sensitive alkenyl moiety. The resulting crude products can be easily purified via standard silica gel column chromatography, yielding materials that meet the stringent purity specifications required for downstream drug development. This level of control over the reaction pathway is critical for R&D teams aiming to establish reproducible and scalable processes for clinical trial materials.
How to Synthesize 3-Alkenyl Quinolin-2(1H)one Efficiently
To implement this synthesis in a laboratory or pilot plant setting, precise adherence to the optimized protocol is essential. The process begins with the careful weighing of the catalyst system components to ensure the correct molar ratios, specifically maintaining a palladium catalyst loading of 10 mol% relative to the substrate. The reaction is conducted in a sealed tube to prevent the escape of any gaseous intermediates and to maintain the necessary pressure for efficient carbonylation. Acetonitrile is selected as the solvent due to its ability to dissolve both the organic substrates and the inorganic bases effectively. Following the reaction period, the workup procedure is straightforward, involving filtration to remove insoluble salts and metal residues, followed by purification. For a detailed, step-by-step breakdown of the exact quantities and procedural nuances, please refer to the standardized synthesis guide below.
- Combine palladium acetate, tris(3-methoxyphenyl)phosphine, molybdenum carbonyl, cesium carbonate, tetrabutylammonium iodide, o-nitrobenzaldehyde, and allyl aryl ether in a sealed tube with acetonitrile.
- Heat the reaction mixture to 100 °C and maintain stirring for approximately 30 hours to ensure complete conversion.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the target 3-alkenyl quinolin-2(1H)one derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical elegance. The primary advantage lies in the substantial cost reduction in pharmaceutical intermediate manufacturing driven by the use of commodity chemicals. O-nitrobenzaldehyde and allyl aryl ethers are commercially available in bulk quantities at relatively low prices compared to specialized organometallic reagents or protected amino acid derivatives often used in alternative routes. Furthermore, the elimination of toxic gases like carbon monoxide from the process input list—replaced by the solid Mo(CO)6—significantly lowers the safety infrastructure costs and regulatory burdens associated with handling hazardous materials. This translates directly into a more resilient and cost-efficient supply chain.
- Cost Reduction in Manufacturing: The economic viability of this process is bolstered by the high catalytic efficiency and the use of inexpensive ligands. Unlike protocols requiring exotic phosphines or high loadings of precious metals, this method utilizes palladium acetate and a simple anisyl-based phosphine, which are cost-effective. Additionally, the reaction achieves high yields, often exceeding 90% for many substrates, which minimizes raw material waste and reduces the cost per kilogram of the final API intermediate. The simplified post-treatment process also reduces labor and solvent consumption during purification, contributing to overall operational expenditure savings.
- Enhanced Supply Chain Reliability: Supply chain continuity is critically dependent on the availability of starting materials. Since o-nitrobenzaldehyde and various substituted allyl aryl ethers are produced on a large industrial scale for other applications, the risk of supply disruption is minimal. The robustness of the reaction conditions means that the process is less sensitive to minor fluctuations in raw material quality, further stabilizing production schedules. This reliability allows manufacturers to maintain consistent inventory levels and meet tight delivery deadlines for their clients in the pharmaceutical sector without the fear of batch failures due to reagent instability.
- Scalability and Environmental Compliance: Scaling this reaction from gram to kilogram or ton scale is feasible due to the absence of extreme pressures or cryogenic temperatures. The use of acetonitrile, a common industrial solvent, facilitates easy recovery and recycling, aligning with green chemistry principles. Moreover, the reduced generation of heavy metal waste and halogenated byproducts simplifies wastewater treatment and disposal compliance. This environmental friendliness not only mitigates regulatory risks but also enhances the corporate sustainability profile of the manufacturing entity, a factor increasingly valued by global pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on the practical aspects of adopting this synthesis route for industrial applications.
Q: What are the key advantages of using o-nitrobenzaldehyde in this synthesis?
A: According to patent CN114478375A, o-nitrobenzaldehyde serves a dual role as both the nitrogen source and the formyl source. This eliminates the need for separate nitrogen-containing reagents and carbon monoxide gas sources, significantly simplifying the reaction setup and reducing raw material costs while maintaining high reaction efficiency.
Q: How does this method handle functional group tolerance compared to traditional routes?
A: The described palladium-catalyzed protocol demonstrates excellent functional group tolerance. It successfully accommodates various substituents on the aryl ring, including halogens (F, Cl), methoxy groups, and trifluoromethyl groups, as well as diverse alkyl and heteroaryl groups on the allyl ether component, yielding products with good to excellent yields up to 99%.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is highly scalable. It utilizes commercially available and inexpensive starting materials like o-nitrobenzaldehyde and allyl aryl ethers. The reaction conditions (100 °C in acetonitrile) are manageable in standard industrial reactors, and the post-treatment involves simple filtration and chromatography, making it viable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Alkenyl Quinolin-2(1H)one Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient synthetic routes in accelerating drug development timelines. Our team of expert chemists has thoroughly analyzed the potential of the palladium-catalyzed reductive aminocarbonylation method described in CN114478375A and is fully equipped to leverage this technology for your projects. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless. Our state-of-the-art facilities are designed to handle complex organometallic reactions with the highest safety standards, and our rigorous QC labs guarantee that every batch of 3-alkenyl quinolin-2(1H)one derivatives meets stringent purity specifications required for clinical and commercial use.
We invite you to collaborate with us to optimize your supply chain for these valuable intermediates. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact our technical procurement team today to request specific COA data for our existing catalog or to discuss route feasibility assessments for your proprietary targets. Let us help you secure a reliable, high-quality supply of these critical building blocks for your next generation of therapeutics.