Scalable Metal-Free Synthesis of Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
Scalable Metal-Free Synthesis of Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
The pharmaceutical industry continuously seeks robust and cost-effective synthetic routes for heterocyclic scaffolds that serve as the backbone of modern therapeutics. A recent technological breakthrough documented in patent CN113105402B introduces a highly efficient preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds, specifically those incorporating valuable trifluoromethyl groups. This innovation addresses critical pain points in API manufacturing by replacing expensive transition metal catalysts with a simple, iodine-promoted system. The significance of this chemical class cannot be overstated, as the 1,2,4-triazole motif is a privileged structure found in numerous blockbuster drugs, including antiviral agents and antidiabetic medications.
The introduction of a trifluoromethyl group into these heterocyclic systems is particularly strategic, as it dramatically enhances the metabolic stability, lipophilicity, and bioavailability of the parent molecule. As illustrated in the structural diversity of known pharmaceuticals below, the versatility of this scaffold supports a wide range of biological activities, making the development of accessible synthetic routes a high priority for R&D teams globally.
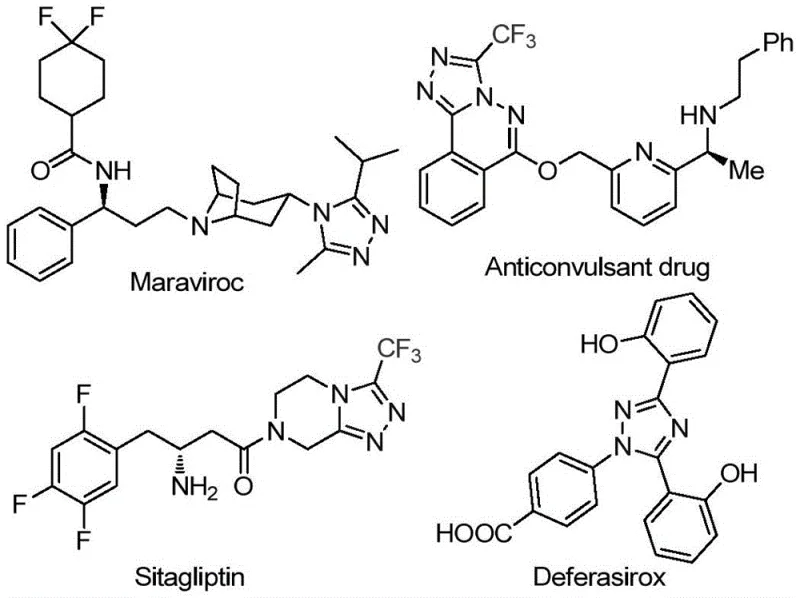
For procurement managers and supply chain directors, the implications of this patent extend beyond mere chemical curiosity. By utilizing cheap and readily available starting materials such as aryl ethyl ketones and avoiding the logistical complexities associated with handling air-sensitive organometallic reagents, this method offers a pathway to cost reduction in pharmaceutical intermediate manufacturing. The ability to operate without strict anhydrous or oxygen-free conditions further lowers the barrier to entry for commercial scale-up, ensuring a more reliable supply chain for these critical building blocks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles, particularly those bearing both acyl and trifluoromethyl substituents, has been fraught with challenges. Traditional methodologies often rely heavily on transition metal catalysts, such as copper or palladium complexes, which introduce significant cost burdens and environmental concerns. The presence of residual heavy metals in the final product is a major regulatory hurdle, necessitating extensive and expensive purification steps to meet the stringent ppm limits required for active pharmaceutical ingredients. Furthermore, many existing protocols require harsh reaction conditions, including high temperatures, strong bases, or strictly inert atmospheres, which complicate reactor design and increase operational risks in a production setting.
Another significant limitation of prior art is the narrow substrate scope. Many conventional methods struggle to tolerate diverse functional groups on the aromatic rings, limiting the chemical space that medicinal chemists can explore during lead optimization. The inability to efficiently introduce specific substituents at the 3, 4, and 5 positions simultaneously often forces researchers to adopt longer, linear synthetic sequences, thereby reducing overall yield and increasing waste generation. These inefficiencies translate directly into higher production costs and longer lead times for getting new drug candidates to market.
The Novel Approach
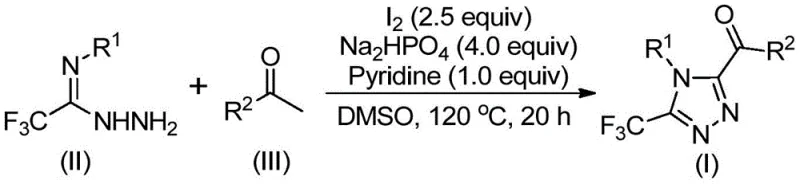
The methodology disclosed in patent CN113105402B represents a paradigm shift by employing a metal-free, iodine-promoted strategy that circumvents the drawbacks of traditional catalysis. This novel approach utilizes elemental iodine and dimethyl sulfoxide (DMSO) to facilitate a tandem oxidation-cyclization sequence. By leveraging the oxidative power of the iodine-DMSO system, the process converts inexpensive aryl ethyl ketones directly into reactive aryl diketone intermediates in situ, which then undergo condensation and cyclization with trifluoroethylimide hydrazide. This telescoped process eliminates the need for isolating unstable intermediates, streamlining the workflow significantly.
Moreover, this method demonstrates exceptional functional group tolerance, allowing for the incorporation of various substituents such as halogens, alkoxy groups, and alkyl chains on both the N-aryl and C-acyl moieties. As shown in the specific examples below, the reaction successfully accommodates electron-rich and electron-deficient substrates, yielding the desired 3,4,5-trisubstituted products with good efficiency. This broad applicability makes it an invaluable tool for generating diverse libraries of triazole derivatives for drug discovery programs.
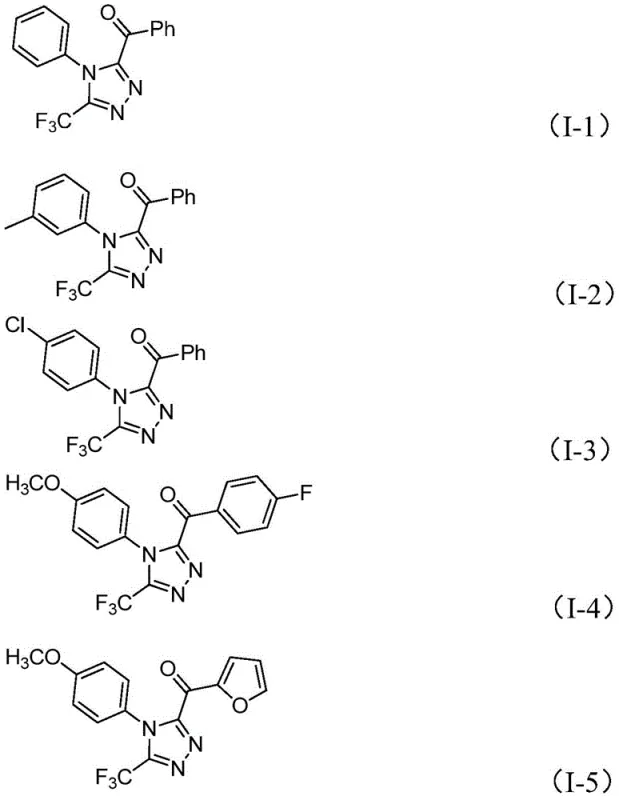
Mechanistic Insights into Iodine-Promoted Tandem Oxidation and Cyclization
Understanding the mechanistic underpinnings of this transformation is crucial for R&D directors aiming to optimize the process for specific substrates. The reaction is believed to proceed through a multi-step cascade initiated by the iodination of the aryl ethyl ketone. In the presence of DMSO, the alpha-iodo ketone intermediate undergoes a Kornblum oxidation to generate an aryl glyoxal (aryl diketone) species. This oxidative step is critical, as it installs the necessary carbonyl functionality required for the subsequent ring closure. The use of DMSO is not merely as a solvent but as an oxygen donor, which is a key feature of this green chemistry approach.
Following the formation of the aryl diketone, the nucleophilic attack by trifluoroethylimide hydrazide occurs, leading to the formation of a hydrazone intermediate. Under the continued influence of iodine and the basic environment provided by pyridine and sodium dihydrogen phosphate, this hydrazone undergoes an intramolecular cyclization. The final aromatization step yields the stable 1,2,4-triazole ring system. The general reaction scheme below elucidates this elegant transformation, highlighting how simple reagents combine to construct a complex heterocyclic architecture with high atom economy.
From an impurity control perspective, this mechanism offers distinct advantages. The absence of transition metals eliminates the risk of metal-catalyzed side reactions such as homocoupling or dehalogenation, which are common pitfalls in cross-coupling chemistry. Furthermore, the reaction conditions are mild enough to preserve sensitive functional groups, minimizing the formation of degradation byproducts. The use of sodium dihydrogen phosphate acts as a buffer, maintaining a pH environment that favors cyclization while preventing the hydrolysis of the imine or hydrazone linkages, thereby ensuring a cleaner crude reaction profile and simplifying downstream purification.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The operational simplicity of this protocol makes it highly attractive for process chemistry teams looking to transfer technology from the lab to the pilot plant. The procedure involves a straightforward two-stage heating process in a single pot, minimizing unit operations and equipment requirements. Detailed standard operating procedures regarding stoichiometry, temperature ramping, and workup protocols are essential for reproducibility. For a comprehensive guide on executing this synthesis with optimal yield and purity, please refer to the standardized steps outlined below.
- Combine aryl ethyl ketone and elemental iodine in dimethyl sulfoxide (DMSO) and heat to 90-110°C for 4-6 hours to initiate oxidation.
- Add additional iodine, sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the mixture to 110-130°C for 12-20 hours to complete the cyclization, followed by filtration and column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, the economic and logistical benefits of this patented method are substantial. By shifting away from precious metal catalysis, manufacturers can decouple their production costs from the volatile pricing of commodities like palladium and copper. This transition not only stabilizes the cost of goods sold (COGS) but also mitigates the geopolitical risks associated with the sourcing of rare earth metals. The reliance on bulk chemicals like iodine and DMSO ensures a robust and resilient supply chain capable of supporting long-term commercial contracts.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts results in direct savings on raw material expenditures. Additionally, the simplified purification process, which avoids the need for specialized scavengers to remove metal residues, reduces the consumption of silica gel and solvents during chromatography. This streamlined workflow translates to lower operational expenses and higher throughput, allowing suppliers to offer more competitive pricing for high-purity pharmaceutical intermediates without compromising on quality standards.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, specifically aryl ethyl ketones and trifluoroethylimide hydrazides, are commodity chemicals that are widely available from multiple global vendors. This abundance prevents supply bottlenecks that often plague specialized reagent-dependent processes. Furthermore, the reaction does not require exotic equipment or extreme conditions such as cryogenic cooling or high-pressure hydrogenation, meaning it can be manufactured in standard glass-lined or stainless steel reactors found in most multipurpose chemical facilities, ensuring consistent delivery schedules.
- Scalability and Environmental Compliance: The protocol is inherently scalable, having been demonstrated to work efficiently from gram scales up to potential tonnage production. The avoidance of heavy metals aligns perfectly with increasingly stringent environmental regulations regarding wastewater discharge and solid waste disposal. By generating less hazardous waste and reducing the environmental footprint of the manufacturing process, companies can achieve better compliance with green chemistry principles, enhancing their corporate sustainability profiles while reducing waste treatment costs.
Frequently Asked Questions (FAQ)
To assist technical decision-makers in evaluating the feasibility of adopting this technology, we have compiled answers to common inquiries regarding the reaction parameters and scope. These insights are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for process development planning.
Q: What are the primary advantages of this iodine-promoted method over traditional transition metal catalysis?
A: This method eliminates the need for expensive and toxic heavy metal catalysts like palladium or copper, significantly reducing raw material costs and simplifying the removal of metal residues to meet stringent pharmaceutical purity standards.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the patent explicitly states that the process is easily scalable from gram levels to industrial quantities, utilizing cheap and readily available starting materials like aryl ethyl ketones without requiring strict anhydrous or oxygen-free conditions.
Q: What is the role of dimethyl sulfoxide (DMSO) in this reaction mechanism?
A: DMSO acts as both the solvent and a critical reactant in the Kornblum oxidation step, facilitating the conversion of aryl ethyl ketones into aryl diketones, which are essential intermediates for the subsequent cyclization with trifluoroethylimide hydrazide.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the pivotal role that advanced heterocyclic intermediates play in the development of next-generation therapeutics. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify identity and assay.
We invite you to collaborate with us to leverage this innovative iodine-promoted synthesis for your specific drug development needs. Whether you require custom synthesis of novel analogs or reliable supply of established intermediates, our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your project volume. Contact us today to request specific COA data and route feasibility assessments, and let us help you accelerate your timeline to market with cost-effective and high-quality chemical solutions.