Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Advanced Pharmaceutical Applications
Scalable Metal-Free Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazoles for Advanced Pharmaceutical Applications
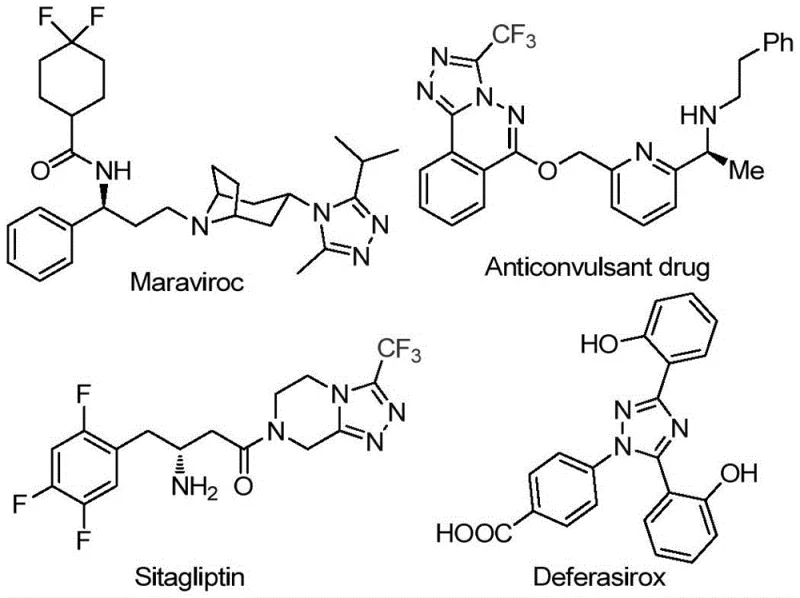
The pharmaceutical and agrochemical industries continuously seek robust, scalable, and environmentally benign synthetic routes for constructing complex heterocyclic scaffolds. Patent CN113105402B introduces a groundbreaking preparation method for 3,4,5-trisubstituted 1,2,4-triazole compounds, a privileged structure found in numerous bioactive molecules. This technology leverages a non-metallic iodine-promoted strategy that circumvents the limitations of traditional transition metal catalysis. By utilizing inexpensive aryl ethyl ketones and trifluoroethylimide hydrazides as starting materials, the process achieves high efficiency under relatively mild conditions. The significance of this innovation lies in its ability to introduce both trifluoromethyl and acyl groups simultaneously into the triazole core, enhancing the lipophilicity and metabolic stability of the resulting drug candidates. For R&D directors and process chemists, this represents a vital advancement in accessing high-value chemical space with reduced operational overhead.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of polysubstituted 1,2,4-triazoles has relied heavily on transition metal catalysts or harsh reaction conditions that pose significant challenges for industrial application. Conventional pathways often require strict anhydrous and oxygen-free environments, necessitating specialized equipment and increasing the overall cost of goods sold (COGS). Furthermore, the reliance on heavy metal catalysts introduces severe complications regarding residual metal limits in final Active Pharmaceutical Ingredients (APIs), demanding expensive and time-consuming purification steps to meet regulatory standards. Many existing methods also suffer from limited substrate scope, failing to tolerate sensitive functional groups or struggling to incorporate trifluoromethyl moieties efficiently. These bottlenecks restrict the rapid iteration required in modern drug discovery and complicate the supply chain continuity for critical intermediates.
The Novel Approach
The methodology disclosed in CN113105402B offers a transformative alternative by employing elemental iodine as a promoter in dimethyl sulfoxide (DMSO). This metal-free approach operates effectively under aerobic conditions, eliminating the need for inert gas protection and simplifying reactor setup. The process utilizes a tandem sequence where aryl ethyl ketones undergo iodination and Kornblum oxidation to form aryl diketones in situ, which subsequently condense with trifluoroethylimide hydrazides. This one-pot strategy not only streamlines the workflow but also ensures excellent atom economy. The reaction conditions are remarkably flexible, accommodating a wide range of substituents on both the hydrazide and the ketone components. This versatility allows for the rapid generation of diverse chemical libraries, making it an ideal platform for lead optimization campaigns in medicinal chemistry.
Mechanistic Insights into Iodine-Promoted Tandem Cyclization
The core of this synthetic innovation lies in the dual role of iodine and DMSO in facilitating oxidative transformation. Initially, elemental iodine interacts with the aryl ethyl ketone in the presence of DMSO to generate an alpha-iodo ketone intermediate, which is further oxidized to an aryl 1,2-diketone via a Kornblum-type oxidation mechanism. This oxidative step is critical as it activates the carbonyl carbon for subsequent nucleophilic attack. Upon the addition of trifluoroethylimide hydrazide, a dehydration condensation occurs to form a hydrazone intermediate. The presence of sodium dihydrogen phosphate and pyridine acts as a buffer system, maintaining the optimal pH for the final cyclization step. Under continued heating at 110-130°C, the hydrazone undergoes intramolecular cyclization promoted by the remaining iodine species, closing the triazole ring and yielding the target 3,4,5-trisubstituted product. This mechanistic pathway avoids the formation of toxic byproducts associated with metal catalysis.
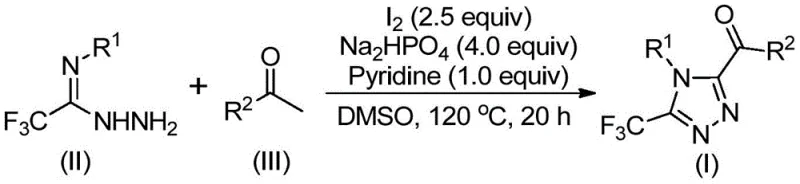
Impurity control is inherently superior in this metal-free system due to the absence of metal-ligand complexes that often degrade into hard-to-remove side products. The primary impurities typically arise from over-oxidation or incomplete cyclization, both of which are easily managed by optimizing the stoichiometry of iodine and the reaction temperature. The use of DMSO as a solvent ensures high solubility for both polar and non-polar intermediates, promoting homogeneous reaction kinetics and minimizing localized hot spots that could lead to decomposition. For quality control teams, this translates to a cleaner crude profile, reducing the burden on downstream purification units. The robustness of the mechanism against moisture and oxygen further enhances batch-to-batch consistency, a critical parameter for GMP manufacturing environments where reproducibility is paramount.
How to Synthesize 3,4,5-Trisubstituted 1,2,4-Triazole Efficiently
The operational simplicity of this protocol makes it highly attractive for process development teams aiming to transfer laboratory methods to pilot plants. The procedure involves a sequential addition strategy where the oxidation phase is completed before introducing the nitrogen source, ensuring that the reactive diketone species is generated in sufficient concentration before cyclization begins. Standard workup procedures involving filtration and silica gel chromatography are sufficient to isolate the product in high purity. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating these results.
- Mix aryl ethyl ketone and elemental iodine in DMSO, heating to 90-110°C for 4-6 hours to initiate Kornblum oxidation.
- Add sodium dihydrogen phosphate, pyridine, and trifluoroethylimide hydrazide to the reaction mixture.
- Heat the solution to 110-130°C for 12-20 hours to complete the cyclization, then purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthesis route offers substantial strategic benefits by decoupling production from the volatility of precious metal markets. The reliance on commodity chemicals like aryl ethyl ketones and elemental iodine ensures a stable and predictable supply chain, mitigating risks associated with geopolitical disruptions affecting rare earth or noble metal availability. The elimination of expensive ligands and metal catalysts directly contributes to significant cost reduction in pharmaceutical intermediate manufacturing. Additionally, the simplified post-reaction processing reduces solvent consumption and waste generation, aligning with green chemistry principles and lowering disposal costs. This efficiency allows suppliers to offer more competitive pricing models while maintaining healthy margins.
- Cost Reduction in Manufacturing: The removal of heavy metal catalysts eliminates the need for specialized scavenging resins and extensive washing protocols, drastically cutting down processing time and material costs. The use of inexpensive reagents like iodine and DMSO, combined with the ability to run reactions under air, reduces utility costs associated with nitrogen blanketing and glovebox operations. These cumulative savings translate into a lower cost per kilogram for the final API intermediate, enhancing the overall profitability of the drug development pipeline.
- Enhanced Supply Chain Reliability: Since the starting materials are widely available commodity chemicals, sourcing is not restricted to single-vendor proprietary supplies. This diversification of the supply base ensures continuity of supply even during market fluctuations. The robustness of the reaction conditions means that production can be easily transferred between different manufacturing sites without requiring bespoke equipment modifications, providing flexibility in capacity planning and inventory management for global supply chains.
- Scalability and Environmental Compliance: The method has been demonstrated to scale effectively from gram to multi-gram levels without loss of yield or selectivity. The absence of toxic heavy metals simplifies wastewater treatment and reduces the environmental footprint of the manufacturing process. This compliance with stringent environmental regulations facilitates faster regulatory approvals and reduces the risk of production shutdowns due to environmental non-compliance, ensuring a steady flow of materials to downstream customers.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this iodine-promoted triazole synthesis. These insights are derived directly from the experimental data and scope defined in the patent documentation, providing clarity for technical decision-makers evaluating this technology for adoption.
Q: What are the key advantages of this iodine-promoted method over traditional heavy metal catalysis?
A: This method eliminates the need for toxic heavy metal catalysts, significantly simplifying post-reaction purification and reducing environmental hazards. It operates under air without strict anhydrous conditions, lowering operational complexity and equipment costs for large-scale production.
Q: Can this synthesis route be scaled for industrial manufacturing of API intermediates?
A: Yes, the patent explicitly demonstrates scalability from gram levels to potential industrial batches. The use of cheap, commercially available starting materials like aryl ethyl ketones and the robust nature of the DMSO solvent system support reliable commercial scale-up.
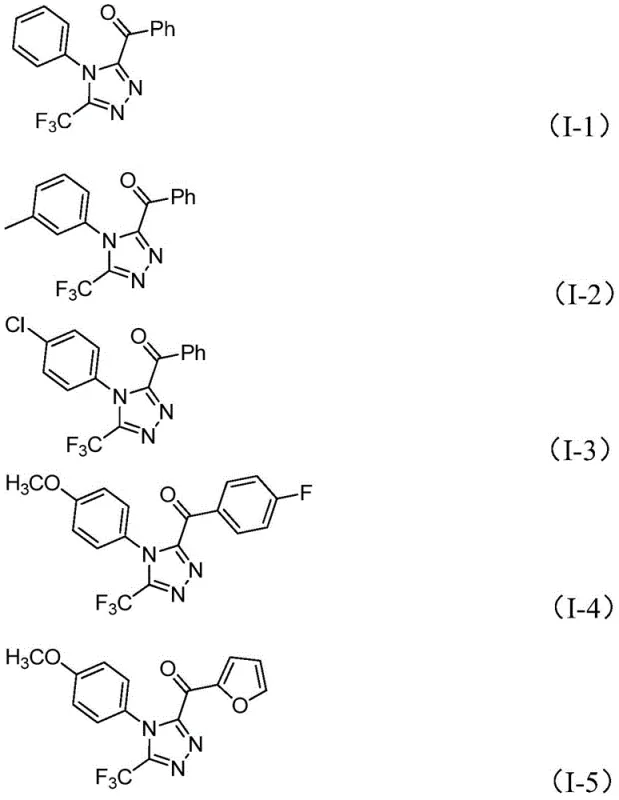
Q: What is the substrate scope for the R1 and R2 groups in this triazole synthesis?
A: The method exhibits broad functional group tolerance. R1 can be various substituted phenyl groups (methyl, methoxy, chloro, trifluoromethyl), while R2 accommodates both substituted phenyl and heteroaryl groups like furan, allowing for diverse library synthesis for drug discovery.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4,5-Trisubstituted 1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the fast-paced pharmaceutical landscape. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries are successfully translated into reliable industrial realities. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 3,4,5-trisubstituted 1,2,4-triazole intermediates meets the highest international standards. Our commitment to quality and consistency makes us a trusted partner for global pharmaceutical companies seeking to optimize their supply chains.
We invite you to collaborate with us to leverage this advanced iodine-promoted technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your drug development timeline while reducing overall production costs.