Advanced Palladium-Catalyzed Synthesis of 2-Trifluoromethyl Quinazolinones for Commercial API Manufacturing
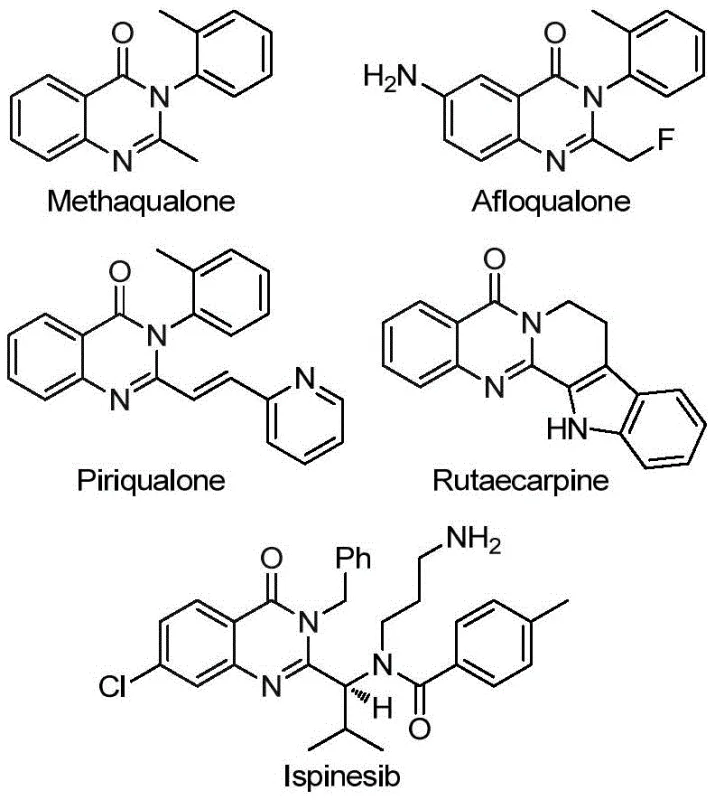
The pharmaceutical industry continuously seeks robust synthetic methodologies for constructing nitrogen-containing heterocycles, particularly quinazolinones, which serve as privileged scaffolds in numerous bioactive molecules ranging from sedatives like Methaqualone to anticancer agents. Patent CN113045503B introduces a groundbreaking preparation method for 2-trifluoromethyl substituted quinazolinone compounds, addressing critical limitations in existing synthetic routes. This innovation leverages a transition metal palladium-catalyzed carbonylation cascade reaction, utilizing readily available trifluoroethylimidoyl chloride and various amines as primary building blocks. The significance of introducing a trifluoromethyl group cannot be overstated, as it significantly enhances the metabolic stability, lipophilicity, and bioavailability of the parent heterocyclic core, making these intermediates highly desirable for modern drug discovery programs. By establishing a protocol that operates under relatively mild thermal conditions with exceptional substrate compatibility, this technology offers a viable pathway for the commercial production of high-value pharmaceutical intermediates.
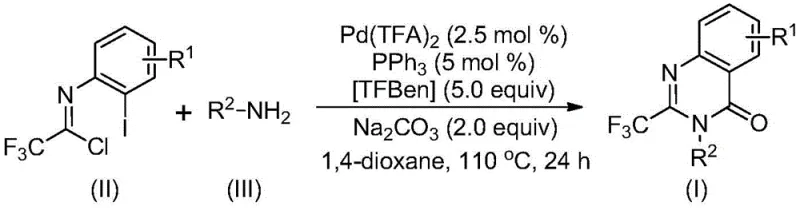
Traditional synthetic approaches for accessing 2-trifluoromethyl quinazolinones have historically been plagued by significant operational and economic inefficiencies that hinder large-scale adoption. Conventional methods often rely on the cyclization of anthranilamides with ethyl trifluoroacetate or trifluoroacetic anhydride, processes that frequently demand harsh reaction conditions, expensive reagents, or tedious pre-activation steps. Furthermore, alternative routes involving isatoic anhydrides or T3P-promoted cascade reactions often suffer from narrow substrate scopes and inconsistent yields, limiting their utility in diverse medicinal chemistry campaigns. In stark contrast, the novel approach detailed in the patent utilizes a palladium-catalyzed system that bypasses these bottlenecks by employing stable trifluoroethylimidoyl chlorides. This method not only simplifies the operational workflow but also dramatically expands the chemical space accessible to chemists, allowing for the introduction of diverse substituents on both the quinazolinone ring and the nitrogen atom without compromising reaction efficiency.
The mechanistic underpinnings of this palladium-catalyzed carbonylation cascade reveal a sophisticated yet efficient catalytic cycle that ensures high conversion rates and product purity. The reaction likely initiates with a base-promoted intermolecular carbon-nitrogen bond coupling to form a trifluoroacetamidine derivative, followed by the oxidative addition of the palladium catalyst into the carbon-iodine bond of the substrate. Crucially, the use of TFBen as a solid carbon monoxide surrogate allows for the safe and controlled release of CO under heating conditions, which then inserts into the carbon-palladium bond to generate a key acyl-palladium intermediate. Subsequent intramolecular cyclization promoted by the base forms a seven-membered ring palladium intermediate, which finally undergoes reductive elimination to release the desired 2-trifluoromethyl-substituted quinazolinone product. This intricate dance of organometallic steps is finely tuned to minimize side reactions, thereby ensuring a clean impurity profile that is essential for downstream pharmaceutical processing and regulatory compliance.
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
Executing this synthesis requires precise attention to reagent stoichiometry and reaction parameters to maximize the yield of the target heterocycle. The process begins by charging a reaction vessel with the palladium catalyst, ligand, base, CO source, and the specific imidoyl chloride and amine substrates in an aprotic organic solvent. The mixture is then subjected to elevated temperatures for an extended period to drive the carbonylation cascade to completion, after which standard workup procedures involving filtration and chromatographic purification are employed. For a comprehensive, step-by-step guide detailing the exact molar ratios, solvent volumes, and purification techniques validated in the patent examples, please refer to the standardized synthesis protocol provided below.
- Combine palladium trifluoroacetate, triphenylphosphine, TFBen, sodium carbonate, trifluoroethylimidoyl chloride, and amine in an organic solvent such as 1,4-dioxane.
- Heat the reaction mixture to 110°C and stir for 16 to 30 hours to allow the carbonylation cascade to proceed to completion.
- Filter the reaction mixture, mix with silica gel, and purify via column chromatography to isolate the target 2-trifluoromethyl-substituted quinazolinone compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, this synthetic methodology offers substantial advantages by fundamentally altering the cost structure associated with producing fluorinated quinazolinone intermediates. The reliance on cheap and commercially available starting materials, such as various amines and trifluoroethylimidoyl chlorides, eliminates the dependency on exotic or custom-synthesized precursors that often carry exorbitant price tags and long lead times. Furthermore, the operational simplicity of the reaction, which avoids the need for high-pressure equipment or cryogenic conditions typically associated with carbonylation reactions, translates directly into reduced capital expenditure and lower energy consumption during manufacturing. These factors collectively contribute to a more resilient supply chain capable of meeting fluctuating market demands without the risk of raw material shortages or production bottlenecks.
- Cost Reduction in Manufacturing: The elimination of expensive pre-activated substrates and the use of a solid CO source instead of hazardous gas cylinders significantly lowers the raw material and safety compliance costs. By streamlining the synthetic sequence into a single pot operation where possible, the process reduces labor hours and solvent usage, leading to substantial overall cost savings in the production of complex heterocyclic intermediates.
- Enhanced Supply Chain Reliability: Since the key reagents like trifluoroethylimidoyl chloride and various amines are widely available from multiple global suppliers, the risk of supply disruption is minimized. This diversification of the supply base ensures continuous production capability, allowing manufacturers to maintain consistent inventory levels and meet tight delivery schedules for downstream API synthesis without being held hostage by single-source vendor limitations.
- Scalability and Environmental Compliance: The reaction conditions are amenable to scale-up from gram to kilogram quantities without significant loss in efficiency, facilitating a smooth transition from process development to commercial manufacturing. Additionally, the use of safer reagents and the generation of manageable waste streams align with green chemistry principles, reducing the environmental footprint and simplifying the regulatory burden associated with waste disposal and emissions control.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented synthesis method, providing clarity for R&D teams evaluating its adoption. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, ensuring accuracy and relevance for process chemists and project managers alike.
Q: What are the advantages of this Pd-catalyzed method over traditional cyclization?
A: Unlike traditional methods requiring harsh conditions or unstable trifluoroacetamides, this method uses cheap, stable starting materials (trifluoroethylimidoyl chloride) and achieves high yields under mild conditions with broad functional group tolerance.
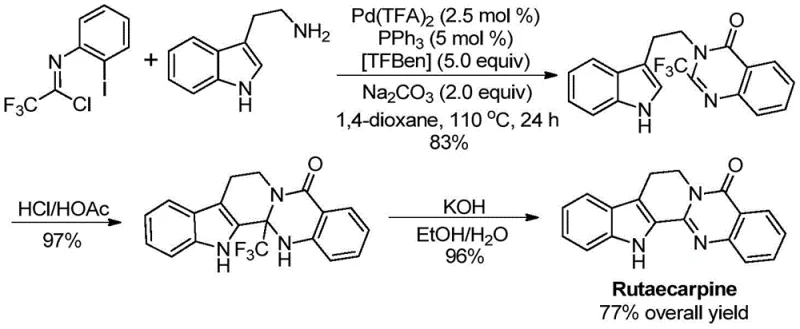
Q: Can this method be applied to synthesize complex drug molecules like Rutaecarpine?
A: Yes, the patent explicitly demonstrates the successful application of this methodology in the efficient total synthesis of Rutaecarpine, achieving a high overall yield through a streamlined three-step sequence.
Q: What is the role of TFBen in this reaction system?
A: TFBen (1,3,5-tricarboxylic acid phenol ester) acts as a solid carbon monoxide substitute, releasing CO in situ under heating conditions to facilitate the carbonylation step without the need for hazardous CO gas cylinders.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that advanced synthetic methodologies play in accelerating drug development timelines and reducing time-to-market for new therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee batch-to-batch consistency and regulatory compliance.
We invite you to collaborate with us to leverage this cutting-edge technology for your specific project needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your supply chain and enhance the economic viability of your drug candidates.