Scalable Synthesis of 2-Trifluoromethyl Quinazolinones Using Iron Catalysis for Global Pharmaceutical Supply Chains
Scalable Synthesis of 2-Trifluoromethyl Quinazolinones Using Iron Catalysis for Global Pharmaceutical Supply Chains
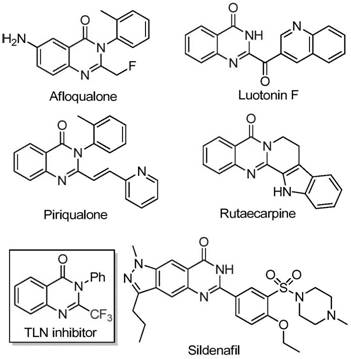
The pharmaceutical industry continuously seeks robust synthetic routes for nitrogen-containing heterocycles due to their prevalence in bioactive small molecules. A pivotal advancement in this domain is detailed in patent CN111675662B, which discloses a highly efficient preparation method for 2-trifluoromethyl substituted quinazolinone compounds. These scaffolds are critical intermediates in the development of anticancer, anticonvulsant, and anti-inflammatory agents, where the introduction of a trifluoromethyl group significantly enhances metabolic stability and lipophilicity. The disclosed methodology leverages a cheap iron catalyst system to overcome the limitations of traditional precious metal catalysis, presenting a compelling value proposition for reliable pharmaceutical intermediate suppliers aiming to optimize their manufacturing portfolios.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted quinazolinones has relied heavily on cyclization reactions involving expensive synthons such as trifluoroacetic anhydride or ethyl trifluoroacetate reacting with anthranilamides or isatoic anhydrides. These conventional pathways are frequently plagued by severe reaction conditions that necessitate rigorous anhydrous environments and elevated pressures, leading to operational hazards and increased energy consumption. Moreover, the substrate scope in these legacy methods is often narrow, failing to accommodate sensitive functional groups without significant degradation or side-reaction formation. The reliance on costly starting materials and the generation of difficult-to-remove impurities result in low overall yields and prohibitive production costs, creating bottlenecks for cost reduction in API manufacturing.
The Novel Approach
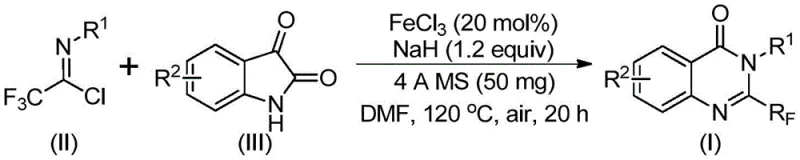
In stark contrast, the innovative route described in the patent utilizes readily available trifluoroethylimidoyl chloride and isatin derivatives as starting materials, catalyzed by inexpensive ferric chloride. This approach fundamentally shifts the economic landscape of production by replacing scarce resources with abundant commodity chemicals. The reaction proceeds through a tandem sequence involving alkali-promoted carbon-nitrogen bond formation followed by iron-catalyzed decarbonylation and cyclization. This mechanistic elegance allows for mild reaction conditions relative to prior art, specifically operating effectively in polar aprotic solvents like DMF. The broad substrate tolerance ensures that diverse analogues can be generated from a single platform, enhancing the designability of the final drug candidates while maintaining high conversion rates.
Mechanistic Insights into FeCl3-Catalyzed Cyclization
The core of this technological breakthrough lies in the dual-activation mechanism facilitated by the iron catalyst and sodium hydride base. Initially, the strong base promotes the nucleophilic attack of the isatin nitrogen onto the imidoyl chloride, forming a transient trifluoroacetamidine intermediate. Subsequently, the ferric chloride acts as a Lewis acid to coordinate with the carbonyl oxygen, facilitating a decarbonylation event that drives the cyclization forward. This iron-mediated pathway avoids the high-energy barriers associated with thermal-only cyclizations, allowing the reaction to proceed at moderate temperatures of 120 °C. The presence of 4A molecular sieves plays a critical role in scavenging trace moisture, which could otherwise hydrolyze the sensitive imidoyl chloride or deactivate the sodium hydride, thereby ensuring consistent reaction kinetics and reproducibility across different batches.
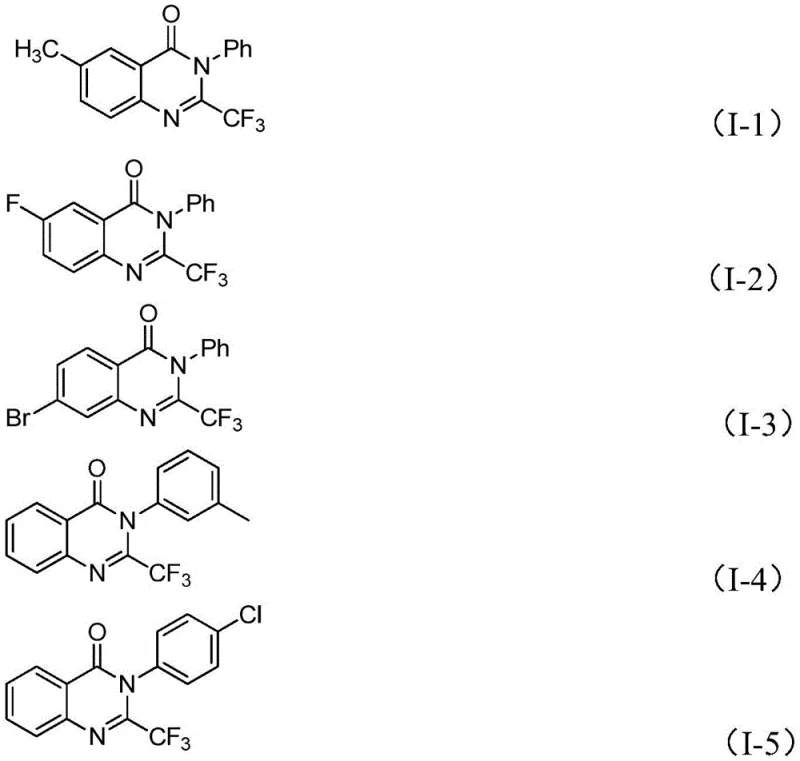
From an impurity control perspective, this mechanism is exceptionally clean. The decarbonylation step releases carbon monoxide gas, which escapes the reaction matrix, driving the equilibrium towards the product and minimizing the formation of polymeric byproducts common in condensation reactions. The high functional group tolerance observed, ranging from electron-rich methoxy substituents to electron-deficient nitro and halogen groups, indicates that the catalytic cycle is robust against competitive coordination or oxidation. This stability is paramount for R&D directors who require high-purity intermediates for downstream biological testing, as it reduces the burden on purification teams and ensures that the final material meets stringent quality specifications without extensive reprocessing.
How to Synthesize 2-Trifluoromethyl Quinazolinone Efficiently
Implementing this synthesis requires precise control over stoichiometry and thermal profiles to maximize yield and safety. The protocol dictates a specific molar ratio of reactants, typically employing a slight excess of the trifluoroethylimidoyl chloride relative to the isatin to drive the reaction to completion. The detailed standardized synthetic steps below outline the exact procedure for combining the iron catalyst, base, and substrates in an organic solvent, followed by the specific heating ramp and workup procedures necessary to isolate the target compound. Adhering to these parameters ensures that the benefits of the novel catalytic system are fully realized in a practical laboratory or pilot plant setting.
- Combine ferric chloride (20 mol%), sodium hydride (1.2 equiv), 4A molecular sieves, trifluoroethylimidoyl chloride, and isatin derivative in anhydrous DMF solvent.
- Stir the reaction mixture initially at 40°C for approximately 10 hours to facilitate initial bond formation, then elevate temperature to 120°C.
- Maintain reaction at 120°C for an additional 20 hours under air atmosphere, followed by filtration and silica gel column chromatography purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this iron-catalyzed methodology represents a strategic opportunity to de-risk the supply of critical heterocyclic building blocks. By eliminating the dependency on volatile and expensive precious metal catalysts, manufacturers can stabilize their raw material costs and insulate themselves from geopolitical fluctuations in the supply of metals like palladium or rhodium. Furthermore, the use of commodity chemicals such as isatins and simple aryl amines ensures a continuous and reliable supply chain, as these precursors are produced globally at massive scales. This shift not only secures the availability of key intermediates but also aligns with broader corporate sustainability goals by utilizing greener, earth-abundant catalysts.
- Cost Reduction in Manufacturing: The replacement of precious metal catalysts with ferric chloride results in a drastic reduction in direct material costs, as iron salts are orders of magnitude cheaper than their noble metal counterparts. Additionally, the simplified workup procedure, which involves basic filtration and standard chromatography rather than complex metal scavenging steps, significantly lowers processing time and labor expenses. The high yields reported, often exceeding 90% for optimized substrates, mean that less raw material is wasted, further driving down the cost per kilogram of the final active pharmaceutical ingredient. This economic efficiency allows for more competitive pricing strategies in the global market without compromising on quality margins.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials such as substituted isatins and aromatic amines mitigates the risk of supply disruptions that often plague specialized reagent markets. Since the reaction tolerates a wide range of functional groups, manufacturers can source alternative grades of substrates if specific lots are unavailable, providing flexibility in vendor selection. The robustness of the reaction conditions, which do not require extreme pressures or cryogenic temperatures, also reduces the likelihood of batch failures due to equipment malfunction, ensuring consistent delivery schedules to downstream customers. This reliability is crucial for maintaining the continuity of drug production lines and meeting strict regulatory deadlines.
- Scalability and Environmental Compliance: The use of DMF as a solvent and iron as a catalyst simplifies the environmental permitting process for scale-up, as iron residues are less toxic and easier to treat in wastewater streams compared to heavy metals. The reaction generates minimal hazardous waste, primarily consisting of inorganic salts that can be disposed of through standard industrial waste channels. The ability to run the reaction under air atmosphere, rather than requiring inert gas blanketing, further reduces operational complexity and infrastructure costs for large-scale reactors. These factors collectively make the commercial scale-up of complex pharmaceutical intermediates more feasible and environmentally sustainable, aligning with modern green chemistry principles.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route. These answers are derived directly from the experimental data and mechanistic understanding provided in the patent documentation, offering clarity on reaction scope, purification, and scalability. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this method for their specific project pipelines.
Q: What are the primary advantages of using FeCl3 over precious metal catalysts for quinazolinone synthesis?
A: The use of ferric chloride (FeCl3) offers a substantial cost reduction compared to traditional palladium or rhodium catalysts. Furthermore, iron is environmentally benign, simplifying waste disposal and regulatory compliance for large-scale API manufacturing.
Q: Does this method tolerate diverse functional groups on the isatin substrate?
A: Yes, the protocol demonstrates excellent functional group tolerance. It successfully accommodates substrates with electron-donating groups like methyl and methoxy, as well as electron-withdrawing groups such as halogens (F, Cl, Br) and nitro groups, yielding products with high purity.
Q: Is the purification process complex for these trifluoromethylated compounds?
A: The post-treatment is streamlined. The reaction mixture can be directly filtered to remove solids, and the crude product is purified via standard silica gel column chromatography, avoiding complex extraction or recrystallization steps often required in older methodologies.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this iron-catalyzed technology for the next generation of therapeutic agents. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to full-scale manufacturing is seamless. Our facilities are equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 2-trifluoromethyl quinazolinone meets the highest international standards for safety and efficacy. We are committed to leveraging our technical expertise to deliver high-purity pharmaceutical intermediates that accelerate your drug development timelines.
We invite you to engage with our technical procurement team to discuss how this novel synthetic route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this iron-catalyzed process for your supply chain. We encourage potential partners to contact us directly to obtain specific COA data and comprehensive route feasibility assessments, allowing you to make informed decisions that drive both scientific success and commercial profitability in the competitive pharmaceutical landscape.