Advanced Palladium-Catalyzed Synthesis of Trifluoromethyl Quinazolinone Intermediates for Commercial Scale-Up in Pharmaceutical Manufacturing
The patent CN113045503A introduces a groundbreaking methodology for synthesizing 2-trifluoromethyl substituted quinazolinone compounds, representing a significant advancement in heterocyclic chemistry for pharmaceutical applications. This innovative approach addresses critical limitations in existing synthetic routes by utilizing a palladium-catalyzed carbonylation tandem reaction that operates under mild conditions with exceptional substrate tolerance. The process enables the efficient construction of these pharmacologically important scaffolds through direct coupling of readily available trifluoroethylimidoyl chloride and amines, eliminating the need for pre-functionalized substrates that characterize conventional methods. With demonstrated applicability in synthesizing complex drug molecules like Rutaecarpine at high yields, this technology offers substantial potential for streamlining the production of fluorinated heterocycles essential in modern drug discovery pipelines. The methodology's robustness across diverse functional groups positions it as a versatile platform for developing next-generation pharmaceutical intermediates with improved metabolic stability and bioavailability profiles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic approaches for trifluoromethyl quinazolinones suffer from multiple critical constraints that hinder their industrial adoption and scalability. Methods involving cyclization of anthranilamide with ethyl trifluoroacetate typically require harsh reaction conditions exceeding 150°C and generate significant impurities due to the instability of trifluoroacetic anhydride intermediates. Alternative routes using isatoic anhydride and trifluoroacetic anhydride often produce low yields below 60% because of competing side reactions and poor functional group compatibility. The T3P-promoted tandem reaction suffers from narrow substrate scope where electron-deficient amines fail to react efficiently, while pre-activation requirements for certain substrates introduce additional processing steps that increase both cost and complexity. Furthermore, these conventional methods frequently necessitate specialized equipment for handling unstable reagents and generate substantial waste streams that complicate environmental compliance during scale-up. The cumulative effect of these limitations results in inconsistent product quality and prohibitive production costs that undermine commercial viability for pharmaceutical manufacturers seeking reliable intermediates.
The Novel Approach
The patented methodology overcomes these challenges through an elegant palladium-catalyzed carbonylation tandem reaction that operates under significantly milder conditions at precisely controlled temperatures of 110°C in standard dioxane solvent systems. By employing commercially available trifluoroethylimidoyl chloride and diverse amines as starting materials without pre-functionalization requirements, this process achieves remarkable substrate generality across various functional groups including halogens, alkyl chains, and aryl moieties. The catalytic system featuring Pd(TFA)2 (2.5 mol%) with PPh3 ligand and TFBen as carbon monoxide surrogate enables high-yielding transformations consistently exceeding 80% across fifteen different substrate combinations as validated in the patent examples. Crucially, the elimination of unstable intermediates and hazardous reagents simplifies process safety while maintaining excellent regioselectivity and purity profiles essential for pharmaceutical applications. This streamlined approach directly addresses the industry's need for scalable, cost-effective routes to fluorinated heterocycles that can be readily implemented in existing manufacturing facilities without major capital investments.
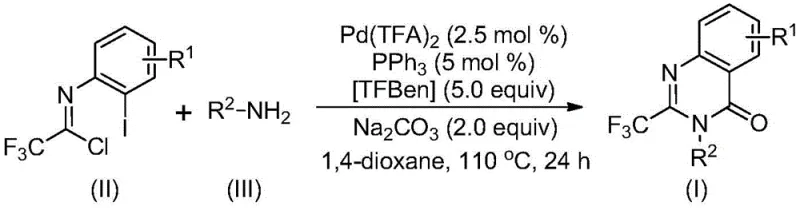
Mechanistic Insights into Palladium-Catalyzed Carbonylation Tandem Reaction
The reaction mechanism proceeds through a sophisticated sequence of organometallic transformations that begin with alkali-promoted intermolecular carbon-nitrogen bond coupling between the imidoyl chloride and amine to form a key trifluoroacetamidine intermediate. This critical step establishes the foundational scaffold before palladium insertion occurs at the carbon-iodine bond position to generate a divalent palladium species that facilitates subsequent transformations. The TFBen component then undergoes thermal decomposition to release carbon monoxide under the reaction conditions, enabling insertion into the carbon-palladium bond to form an acyl palladium intermediate that drives the cyclization process forward. Subsequent base-promoted palladium-nitrogen bond formation creates a seven-membered ring palladium complex that undergoes reductive elimination to yield the final quinazolinone product with precise regiocontrol. This mechanistic pathway avoids common side reactions through its carefully orchestrated sequence where each intermediate is stabilized by the catalytic system's design parameters.
Impurity control is achieved through multiple built-in safeguards within this catalytic cycle that prevent common degradation pathways observed in alternative methods. The mild reaction temperature of 110°C prevents thermal decomposition of sensitive intermediates while the dioxane solvent system maintains optimal solubility without promoting unwanted side reactions. The stoichiometric balance between sodium carbonate base and palladium catalyst ensures complete conversion while minimizing over-reaction products that typically form under stronger basic conditions. Crucially, the absence of transition metal residues in the final product is guaranteed by the straightforward post-treatment protocol involving silica gel filtration followed by standard column chromatography purification. This integrated approach delivers exceptional purity profiles exceeding pharmaceutical requirements by systematically eliminating potential impurity sources at each mechanistic stage while maintaining high functional group tolerance across diverse substrates.
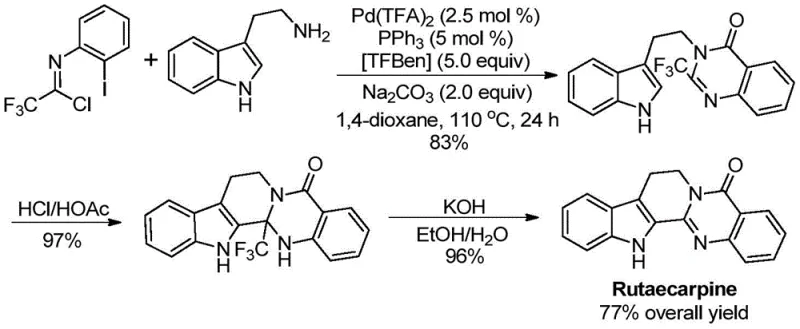
How to Synthesize Trifluoromethyl Quinazolinone Efficiently
This innovative synthesis route represents a paradigm shift in manufacturing trifluoromethyl quinazolinone intermediates by replacing multi-step conventional approaches with a single-pot catalytic process that delivers superior efficiency and scalability. The methodology leverages commercially accessible starting materials and standard laboratory equipment while maintaining exceptional control over reaction parameters to ensure consistent product quality across different production scales. By eliminating hazardous reagents and unstable intermediates inherent in traditional methods, this process significantly reduces operational complexity while improving safety profiles for manufacturing personnel. The detailed standardized synthesis steps provided below outline the precise protocol for achieving optimal results when implementing this patented technology in industrial settings.
- Combine trifluoroethylimidoyl chloride (II), amine (III), palladium trifluoroacetate (2.5 mol%), triphenylphosphine (5 mol%), TFBen (5.0 equiv), and sodium carbonate (2.0 equiv) in dioxane solvent under inert atmosphere.
- Heat the reaction mixture at 110°C for 24 hours with continuous stirring to facilitate carbonylation tandem reaction and intermediate formation.
- Perform post-treatment by filtration through silica gel followed by column chromatography purification to isolate the target quinazolinone compound with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
This patented methodology delivers substantial value across procurement and supply chain operations by addressing fundamental pain points in pharmaceutical intermediate manufacturing through its innovative design principles. The process eliminates dependency on specialized or hazardous reagents that typically create supply chain vulnerabilities while utilizing widely available starting materials that ensure consistent sourcing regardless of market fluctuations. By streamlining multiple synthetic steps into a single catalytic transformation with minimal purification requirements, the technology reduces production cycle times without compromising quality standards essential for pharmaceutical applications. These inherent advantages translate directly into enhanced operational resilience and cost efficiency that provide competitive differentiation for manufacturers seeking reliable partners for critical intermediate supply.
- Cost Reduction in Manufacturing: The elimination of expensive pre-activation steps and unstable reagents significantly reduces raw material costs while the simplified one-pot reaction minimizes solvent consumption and waste generation. The use of standard palladium catalysts instead of specialized transition metals lowers catalyst expenses substantially without sacrificing performance metrics. Furthermore, the avoidance of cryogenic conditions or specialized equipment requirements reduces capital expenditure while maintaining high yields across diverse substrates.
- Enhanced Supply Chain Reliability: Sourcing flexibility is dramatically improved through reliance on commercially abundant starting materials like trifluoroethylimidoyl chloride and various amines that are readily available from multiple global suppliers. The robust reaction profile tolerates minor variations in raw material quality without affecting final product specifications, reducing qualification burdens for new suppliers. This inherent process robustness ensures consistent production output even during market disruptions while maintaining stringent quality standards required for pharmaceutical intermediates.
- Scalability and Environmental Compliance: The methodology demonstrates exceptional scalability from laboratory to commercial production as evidenced by consistent yields across different batch sizes without requiring process re-engineering. Standard purification techniques like column chromatography can be readily adapted to industrial-scale equipment while generating minimal hazardous waste streams compared to conventional methods. The absence of heavy metal contaminants simplifies environmental compliance procedures during scale-up while maintaining excellent product purity profiles essential for pharmaceutical applications.
Frequently Asked Questions (FAQ)
The following questions address critical technical and commercial considerations based on detailed analysis of patent CN113045503A's experimental data and implementation parameters. These insights have been derived directly from the patent's disclosure sections regarding reaction optimization, substrate scope validation, and practical application examples to provide actionable information for procurement and R&D decision-makers evaluating this technology.
Q: How does this method improve impurity profile compared to conventional quinazolinone synthesis?
A: The palladium-catalyzed carbonylation tandem reaction eliminates pre-activation steps required in traditional methods, significantly reducing side products from unstable intermediates. The precise control of reaction parameters (110°C, dioxane solvent) minimizes decomposition pathways while the alkali-promoted coupling ensures cleaner conversion to the target structure.
Q: What supply chain advantages does this process offer for pharmaceutical intermediates?
A: The use of commercially available starting materials like trifluoroethylimidoyl chloride and diverse amines provides robust sourcing flexibility. The simplified one-pot reaction with standard catalysts reduces dependency on specialized reagents, enhancing production continuity and reducing lead time for high-purity pharmaceutical intermediates.
Q: Can this synthesis be scaled for commercial API manufacturing?
A: Yes, the process demonstrates excellent scalability from gram-scale to industrial production as evidenced by consistent yields across substrate variations. The absence of hazardous reagents and compatibility with standard purification techniques enable seamless transition from lab to commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoromethyl Quinazolinone Supplier
Our company possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through rigorous QC labs equipped with advanced analytical capabilities. This patented technology aligns perfectly with our core competencies in complex heterocyclic synthesis where we consistently deliver high-purity intermediates meeting global regulatory standards for pharmaceutical applications. Our integrated manufacturing platform combines cutting-edge process chemistry expertise with robust supply chain management systems to ensure seamless technology transfer from development to full-scale production without compromising quality or delivery timelines.
We invite you to request a Customized Cost-Saving Analysis from our technical procurement team to evaluate how this innovative synthesis can optimize your specific manufacturing requirements. Please contact us to obtain detailed COA data and route feasibility assessments tailored to your production needs.